LE TAPPE STORICHE DELL’IMMUNOLOGIA
di Sergio Barocci – Università di Genova per la terza età e divulgatore scientifico.
dagli anni sessanta DEL NOVECENTO ai giorni nostri (PARTE III)

Gli anni ‘60 e gli anni ‘70 rappresentarono qualcosa di nuovo rispetto al passato. Entrambi i decenni segnarono l’inizio di una tappa più avanzata dell’immunologia sia umorale che cellulare, rappresentando un periodo nel quale si incominciarono a chiarire non solo le basi molecolari della risposta immunitaria, ma anche a comprendere i meccanismi che provocavano le malattie, con nuove prospettive diagnostiche e terapeutiche.
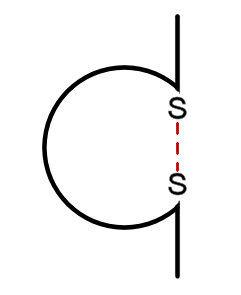
È agli inizi degli anni ’60 che vennero avviati studi sulle immunoglobuline che in pochi anni portarono alla scoperta della struttura chimica e dell’eterogeneità degli anticorpi. Queste ricerche di fondamentale importanza per l’immunologia e per i risvolti clinici furono condotti dagli immunochimici Rodney Robert Porter, Gerald Maurice Edelman ed Alfred Nisonoff.
struttura base della molecola di un anticorpo

È al lavoro di questi ricercatori che si deve attribuire il merito della comprensione della struttura base della molecola di un anticorpo consistente in due catene polipeptidiche leggere L e in due pesanti H, composte di regioni costanti (C) e di regioni variabili (V).
Ciascuna catena L è legata ad una catena H da un ponte disolfuro così come le catene H sono legate tra loro da almeno un ponte disolfuro. Essi dimostrarono inoltre come l’anticorpo poteva poi essere tagliato enzimaticamente in due frammenti Fab che legavano l’antigene ma che non formavano un precipitato e in un frammento Fc che invece mediava le funzioni effettrici quali il legame con il sistema complementare.
Successive analisi cristallografiche ai raggi X fornirono un’ulteriore prova che i siti di legame per l’antigene erano formati da tre segmenti ipervariabili nella catena L e da tre nella catena H (le 6 regioni che determinano la complementarietà o CDR) e che esistevano 5 tipi di catene pesanti differenti per caratteristiche chimico-fisiche, identificate con le lettere dell’alfabeto greco α, δ, ε, γ e μ. Sulla base della tipologia di catena pesante vennero identificate le diverse classi di immunoglobuline denominandole con la corrispondente lettera dell’alfabeto latino: IgA, IgD, IgE, IgG e IgM. Queste vennero poi ulteriormente suddivise in sottoclassi (quattro per le IgG e due per le IgA).
Dominio delle immunoglobuline
Nel 1969 G.M. Edelman presentò la prima sequenza amminoacidica completa di un anticorpo introducendo il concetto di dominio per indicare la regione globulare compatta di una catena pesante o leggera che presenta una struttura terziaria coerente. Egli dimostrò che:
• la regione variabile della catena leggera L (VL) era simile per sequenza alla regione variabile della catena pesante H (VH);
• la regione costante della catena pesante (CH) era costituita da tre parti di uguali dimensioni (CH1,CH2,CH3) simili anche in sequenza;
• la regione costante della catena leggera (CL) era simile ai tre domini della regione costante:
• un ponte disolfuro intracatena si trovava nella stessa posizione in ciascun dominio di entrambe le catene L e H.
Tutte queste somiglianze suggerirono che gli anticorpi o immunoglobuline si ripiegassero in domini compatti, ciascuno dei quali deputato ad una funzione molecolare caratteristica nel caso di formazione di complessi antigene-anticorpo (attivazione cascata complementare, attivazione della fagocitosi e rilascio di granuli da parte dei mastociti).
ANTICORPI ED ANTIGENI

Successivi studi cristallografici rivelarono il modo in cui gli anticorpi erano in grado di legare apteni e antigeni a fessure, a tasche e ad estese superfici piatte formate dai CDR e che l’insieme di interazioni utilizzate per ottenere la complementarietà e l’alta affinità era simile a quello impiegato nella formazione dei complessi enzima-substrato. La scoperta di regioni variabili e regioni costanti distinte nelle catene L e H sollevò la possibilità che i geni delle immunoglobuline avessero un’architettura insolita.
Già nel 1965 William Dreyer e Claude Bennet proposero che i geni multipli V fossero separati da un singolo gene C nel DNA embrionale. Secondo il loro modello, uno di questi geni V veniva unito al gene C durante il differenziamento cellulare che produceva anticorpi. Per verificare tale ipotesi si dovettero aspettare diversi anni.
La tecnica del DNA ricombinante e l’uso di sonde genetiche fornirono a Susumu Tonegawa gli strumenti tecnici per analizzare la diversità anticorpale durante la sintesi citoplasmatica. A partire dal 1976, egli scoprì il fenomeno del meccanismo di rimescolamento genetico, denominato “riarrangiamento genico” delle cellule per la diversità anticorpale, utilizzato dai linfociti B per la produzione degli anticorpi specifici.
Da Kohler e Milstein ai giorni nostri

Verso la metà degli anni sessanta divenne chiaro come la risposta immunitaria fosse la risultante di una collaborazione fra i linfociti maturati nel timo (linfociti T) e quelli provenienti dal midollo osseo (linfociti B). Per comprendere il ruolo svolto dall’immunità cellulare nella risposta immunitaria si dovette aspettare il 1961, anno in cui venne scoperto da Jacques Francis Albert Pierre Miller (2aprile 1931 – ) il ruolo svolto dal timo nella produzione dei linfociti T grazie ad esperimenti di timectomia nel topo. Miller, lavorando presso l’Istituto Walter e Eliza Hall of Medical Research di Melbourne in Australia, dimostrò che alcuni topi privi del timo non rigettavano innesti di cute proveniente da altri topi: da qui la supposizione che dal timo dovessero avere origine le cellule responsabili del rigetto.
A quel tempo, si riteneva che gli anticorpi derivassero dai linfociti ma non si conosceva ancora quale legame esistesse tra i linfociti che producevano anticorpi e quelli invece coinvolti nel rigetto dei trapianti.
LINFOCITI E LINFOCITI

In questo contesto si trovò ad operare Max Dale Cooper che intuì che i linfociti produttori di anticorpi e i linfociti timo-derivati che conducevano al rigetto, non erano i medesimi.
Un ulteriore indizio arrivò da uno studio condotto nel 1956 da Bruce Glick e Timothy Chang sulla scoperta fatta nei polli di un particolare organo “la borsa di Fabrizio” sospettato di giocare un importante ruolo nella produzione degli anticorpi; una volta rimossa la borsa di Fabrizio i livelli anticorpali nei polli risultavano infatti bassissimi.
M. Cooper e Robert Good (21maggio 1922 – 13giugno 2003) confermarono nel 1965 i risultati degli studi del 1956: polli infettati con batteri del genere Brucella e sottoposti a radiazioni il giorno dopo essere stati privati della borsa di Fabrizio, non erano in grado di produrre anticorpi anti-Brucella.
LINFOCITI B E LINFOCITI T

Questo dato indusse a pensare che esistevano due classi di linfociti con cui l’organismo si difendeva dagli attacchi esterni e che questi svolgessero funzioni differenti.
I linfociti B derivanti dalla borsa di Fabrizio erano necessari per la risposta anticorpale umorale mentre i linfociti T (timo dipendenti) mediavano reazioni di ipersensibilità ritardata come nei casi di rigetto.
Rimaneva da chiarire quale era l’equivalente umano della borsa di Fabrizio. La risposta a tale quesito arrivò nel 1974: l’equivalente nei mammiferi della borsa di Fabrizio dei polli era il tessuto ematopoietico da cui prendevano origine tutte le cellule del sangue, comprese quelle che sarebbero state identificate successivamente come linfociti B. Fondamentale a tale riguardo fu anche il contributo dato da Henry Claman che nel 1966 identificò le cellule CFA (cellule che formano anticorpi) come prodotto dei linfociti B.
APOPTOSI CELLULARE

Agli inizi degli anni ‘70 fu descritto il fenomeno dell’apoptosi cellulare cioè quel processo mediante il quale le cellule in sovrannumero o danneggiate e non riparabili nonostante l’intervento di proteine chiamate HSP (Heat Shock Protein) programmano la loro morte tramite una molecola di superficie cellulare detta Fas (appartenente alla famiglia dei TNF) e del suo ligando FasL.
A metà degli anni ‘70 venne individuato un gruppo di linfociti del sangue periferico detti cellule “null” in quanto non esprimenti le molecole di membrana caratteristiche dei linfociti T e B; a queste cellule, dall’aspetto granulare e con la funzione ben definita di difendere l’ospite nei confronti di cellule tumorali, sarebbe stato dato il nome di ”linfociti natural killer” (NK).
Un’ulteriore accelerazione nella comprensione dei meccanismi immunologici ebbe luogo nel 1975, grazie all’impiego di metodiche di biologia molecolare da parte di Niels Kaj Jerne, Georges J. Köhler e César Milstein per la produzione di “anticorpi monoclonali”.
ANTICORPI MONOCLONALI

Questi ricercatori scoprirono che si potevano ottenere grandi quantità dello stesso anticorpo, e con qualsiasi specificità desiderata, mediante la fusione di una cellula produttrice di anticorpo ricavata dalla milza di ratti o conigli immunizzati per un antigene noto, con una cellula trasformata della linea B (mielomatosa).
L’invenzione degli ibridomi, ovvero di cellule ingegnerizzate per produrre anticorpi monoclonali, ha permesso di compiere enormi progressi nel campo della ricerca, delle applicazioni diagnostiche per immagini e in quello terapeutico.
Tra il 1973 e il 1975 vennero definite le classi di antigeni codificate dal Complesso Maggiore di Istocompatibilità e venne approfondito il ruolo del rapporto fra aptene e carrier nell’ambito dei meccanismi di stimolo e soppressione.
Il Complesso Maggiore di Istocompatibilità

Le conoscenze acquisite nel corso di questi anni sul controllo genetico della risposta immunitaria adattativa da parte dei geni collegati al sistema genetico MHC ad opera di Hugh O’Neill McDevitt e di Baruj Benacerraf, rappresentarono il punto di partenza per altri studi sulla restrizione genica nelle interazioni cellulari con la dimostrazione del fenomeno della restrizione del sistema MHC nel riconoscimento antigenico dei linfociti T, ovvero che i linfociti T helper CD4+ riconoscono solo molecole MHC di classe II, mentre i linfociti CD8+ solo le molecole MHC di classe I e solo con il legame al complesso peptide-MHC e non con il solo peptide, essi possono innescare la risposta immunitaria.
Benacerraf scoprì che la risposta immunitaria nei confronti dell’antigene che stava utilizzando era controllata da un singolo gene autosomico dominante e, successivamente, ipotizzò che i geni della risposta immunitaria (detti IR da Immune Response) fossero implicati nel riconoscimento dell’antigene.
teoria del “network idiotipico”

Oggi sappiamo che i geni IR sono i medesimi geni dell’MHC che codificano per le molecole MHC o HLA nell’uomo.
Nel 1974 N. K. Jerne postulò invece la teoria del “network idiotipico”, un complesso sistema di regolazione interna fondato sulla mediazione tra recettori di membrana e anticorpi con ‛idiotipi‘ complementari. Nasceva con lui l’immagine di un “sistema immunitario” costantemente attivato da segnali umorali e cellulari, positivi e negativi, modulati in funzione della qualità della risposta finale che doveva essere prodotta.
Durante gli anni ’70 furono anche intrapresi numerosi studi per individuare la struttura chimica delle molecole MHC sia nell’uomo che nel topo. Si scoprì che le molecole MHC di classe I erano formate da una catena pesante α e da una catena leggera β, legata in maniera non covalente con la porzione extracellulare della catena α, chiamata β2-microglobulina e presente su quasi tutte le cellule nucleate, mentre la molecola MHC di classe II era invece formata da due catene pesanti, α e β associate non covalentemente ed espresse solo su linfociti B, macrofagi, cellule dendritiche e cellule endoteliali.
Cluster of Differentiation

Le cellule che esprimevano MHC sulla loro superficie e che erano in grado di processare gli antigeni peptidici estranei “non self”, vennero chiamate cellule presentanti l’antigene o APC (differenti tipi di cellule dendritiche, macrofagi tissutali attivati, monociti e cellule B attivate).
Nel 1982, durante il 1° Workshop Internazionale sugli antigeni di differenziazione leucocitari, si posero le basi per un sistema uniforme di nomenclatura per le molecole di membrana leucocitarie, con la descrizione dei markers cellulari di superficie su linfociti, macrofagi e cellule endoteliali riconosciuti da determinati anticorpi monoclonali e l’adozione del sistema di denominazione “CD” (Cluster of Differentiation).
Nel 1987 Jack Leonard Strominger e Don Craig Wiley evidenziarono la struttura tridimensionale delle molecole MHC di classe I e II con l’impiego della cristallografia a raggi X.
struttura tridimensionale delle molecole MHC

Nello stesso anno, sempre tramite la cristallografia a raggi X, individuarono in queste molecole la regione di legame dei peptidi (un solco profondo fiancheggiato da alfa eliche) presentati sulla superficie cellulare dei linfociti in modo altamente diversificato.
Risultò infatti che ogni molecola MHC conteneva una tasca posta nella porzione extracellulare e una coppia di domini immunoglobulinici o Ig legati alla membrana cellulare. La tasca, costituita da due ά-eliche appoggiate su un foglietto-β costituito da 8 filamenti, rappresentava la regione che conteneva gli amminoacidi polimorfici che determinavano le differenze fra MHC.
I domini Ig, che non erano polimorfi, contenevano il sito di legame per i recettori dei linfociti T.
linfociti T citotossici

Rolf Martin Zinkernagel e Peter Charles Doherty scoprirono che i linfociti T citotossici erano in grado di uccidere una cellula infettata da un virus solo se questa condivideva l’antigene di istocompatibilità MHC di classe I, mentre altri ricercatori stabilirono che le molecole MHC di classe II erano necessarie per la cooperazione cellulare fra linfociti T e B.
In particolare, l’antigene timo-dipendente doveva essere presentato dalle cosiddette cellule accessorie insieme agli antigeni di classe II per essere riconosciuto dai linfociti T helper CD4+ [è del 1986 la differenziazione dei T helper (Th) in due distinte sottopopolazioni, rispettivamente in Th1, che regolano la risposta cellulo-mediata contro le infezioni intracellulari e in Th2, coinvolti nella lotta contro le infezioni associate all’immunità anticorpo-mediata].
In base a questo approccio centrato sul recettore dei linfociti T, gli antigeni esterni venivano riconosciuti e quindi attivavano una risposta immunitaria, essendo in grado di modificare le molecole di istocompatibilità che ovviamente dovevano appartenere allo stesso genotipo della cellula T.
riconoscimento dell’antigene esterno

Valeva a dire che l’antigene era rielaborato dalle cellule e presentato sulla superficie nel contesto delle molecole di istocompatibilità e, dunque, i linfociti T riconoscevano come estraneo questo “self modificato”.
Tale capacità di riconoscere le molecole di istocompatibilità autologhe era appresa dai linfociti T durante la loro maturazione nell’ambiente timico, attraverso un processo selettivo, positivo e negativo, che lasciava sopravvivere solo quelle cellule che trasportavano recettori con una propensione a riconoscere gli antigeni di istocompatibilità autologhi in congiunzione con peptidi estranei e che, però, erano tolleranti verso i propri componenti.
Verso la metà degli anni ’80 venne approfondito il ruolo del rapporto fra aptene e carrier nell’ambito dei meccanismi di stimolo e soppressione ed incominciarono ad essere caratterizzati molecolarmente anche i diversi tipi di recettori cellulari coinvolti nel processo di riconoscimento antigenico MHC-correlato.
T cell receptor

Risale agli anni 1982 e 1983 l’identificazione del T cell receptor (TRC) responsabile del riconoscimento degli antigeni presentati dal complesso maggiore di istocompatibilità (MHC). Questo fu possibile attraverso l’utilizzo di anticorpi monoclonali prodotti verso diversi cloni di linfociti T da parte di James Allison, Philippa Marrack e John W. Kappler. Questi anticorpi furono indispensabili per individuare i diversi cloni (clone –specifico o clonotipico) così come la loro struttura molecolare espressione di riarrangiamenti genici.
Allison scoprì che i TCR erano degli eterodimeri costituiti da una catena ά e una catena β (TCR ά e TCR β) o da una catena γ e una δ (TCR γ e TCR δ) permettendo una distinzione tra linfociti T αβ e linfociti T γδ e che queste catene, analogamente alle catene pesanti e leggere delle immunoglobuline, possedevano regioni costanti e variabili.
Complementarity-Determining Regions

All’interno della regione variabile si distinguevano tre siti ipervariabili che corrispondevano ad anse all’interno di una struttura organizzata in foglietti β: queste regioni erano le CDR (Complementarity-Determining Regions) del TCR. Sempre Allison dimostrò che il complesso del TCR era costituito dal TCR stesso e da proteine deputate alla trasduzione del segnale legate tramite legami non-covalenti mentre furono S. M. Hedrick e M. M. Davis nel 1984 a descrivere sia l’organizzazione multigenica che il riarrangiamento dei geni del TCR; quest’ultimo con meccanismi simili a quelli che avvenivano per il riarrangiamento del DNA germinale delle immunoglobuline.
Intorno alla metà degli anni ‘60 si era osservato che i surnatanti di colture leucocitarie miste contenevano fattori, prodotti dai linfociti, dotati di attività biologiche. Lo studio di diversi sistemi sperimentali aveva dimostrato che l’azione di tali fattori poteva essere sia di tipo inibitorio sia di tipo stimolatorio e poteva interessare sia le cellule dell’immunità specifica sia quelle dell’immunità aspecifica.
LE LINFOCHINE E CITOCHINE

Nel 1969 i fattori responsabili di tali fenomeni furono chiamati “linfochine”.
L’anno successivo si dimostrò che l’attività biologica di un fattore derivante da cellule non linfocitarie era in grado di stimolare i linfociti, per tale ragione denominato LAF (Lymphocite Activating Factor).
La scoperta che i fattori umorali in grado di amplificare o modificare le risposte immunitarie potevano essere prodotti anche da cellule non linfoidi indusse l’immunologo britannico Stanley Cohen a proporre nel 1974 di denominare questi mediatori, nel loro insieme, “citochine”.
In un breve lasso di tempo si assistette alla scoperta di numerosi fattori dotati di attività biologica e di pari passo all’utilizzo di numerosi acronimi utilizzati per indicare tali attività; ben presto risultò evidente che alcuni fattori conosciuti con nomi diversi corrispondevano alla stessa citochina, che agiva diversamente a seconda del contesto.
PROLIFERAZIONE DELLE CELLULE T IN COLTURA

Nel 1976 si scoprì la possibilità di far proliferare le cellule T in coltura in terreno precedentemente condizionato da cellule T attivate con un mitogeno.
Alla fine degli anni ’70 si attribuì questa proprietà a un fattore che inizialmente fu chiamato fattore di crescita delle cellule T (TCGF, T cell growth factor), destinato a diventare la citochina più famosa, sia perché la prima ad essere descritta da un punto di vista biochimico, sia perché intorno ad essa si crearono nella seconda metà degli anni ’80 diverse aspettative terapeutiche. Questa citochina è oggi conosciuta con il nome di interleuchina-2 (IL-2), mentre il LAF è oggi noto come IL-1.
Tra il 1983 e il 1984 Tadatsugu Taniguchi focalizzò le sue ricerche sull’immunità e sull’oncogenesi, in particolare sui meccanismi di trasduzione del segnale e sull’espressione genica di alcune citochine. Identificò oltre alla sequenza del DNA codificante per la citochina IL-2 anche quelli codificanti per l’interferone-beta (IFN-β) e i fattori di trascrizione (IRF) della cellula verso le infezioni virali.
più di 100 citochine

In pochi anni furono identificate più di 100 citochine (implicate nelle interazioni cellulari e nell’infiammazione dei tessuti) e numerose “chemochine” implicate invece nell’attivazione e nel reclutamento (chemiotassi) dei leucociti nei siti di flogosi, delle molecole di adesione (IgSF CAMs, selettine, integrine, caderine) e nuove famiglie di fattori di trascrizione oltre che acquisite numerose informazioni sul funzionamento della rete immunitaria cellulare.
Negli anni ’90 si assistette ad una svolta nelle conoscenze del funzionamento del nostro sistema immunitario: particolare attenzione venne rivolta al sistema innato con la scoperta di un tipo di cellule che per la loro struttura ramificata erano state chiamate dendritiche tra gli anni ’70 e ’80 da Ralph Steinman e a particolari strutture molecolari associate ai patogeni detti PAMPS (Pathogen Associated Molecular Patterns) riconosciute da alcuni recettori denominati TLR (Toll-Like Receptors) espressi in molti tipi cellulari tra cui le cellule dendritiche.
Il gene Toll

Nel 1996 Jules Hoffmann, studiando il moscerino della frutta (Drosophila Melanogaster) con una mutazione del gene Toll (“notevole” in tedesco), gene coinvolto nello sviluppo embrionale, (identificato nel 1985 nella Drosophila da Christiane Nüsslein-Volhard) scoprì che tale gene era coinvolto nel riconoscimento di patogeni e che la sua attivazione era necessaria al sistema immunitario.
Due anni dopo e precisamente nel 1998, Bruce Beutler studiando un ceppo di topi resistente allo shock termico scoprì che anche questi possedevano un gene mutato molto simile al gene Toll della Drosophila e in grado di codificare un recettore TLR capace di legare una tossina LPS prodotta dai batteri e responsabile dello shock termico. Questa tossina entrando nella tasca del TLR attivava il processo infiammatorio scatenando la risposta immunitaria.
LE CELLULE DENDRITICHE

Queste ricerche posero le basi per comprendere che le cellule dendritiche rappresentavano il collegamento tra le due risposte immunitarie, perchè da una parte capaci di captare i segnali della risposta immunitaria innata e dall’altra di attivare la risposta immunitaria adattativa in quanto attivavano i linfociti T. Inoltre, si comprese che il sistema innato e quello adattativo risultavano attivamente connessi, al punto tale che il sistema adattativo non poteva manifestarsi senza l’attivazione del sistema innato.
Nel 1992 C. Benoist e D. Mathis scoprirono che era la parte esterna della catena del TCR a cambiare da linfocita a linfocita; nel 1994 S. Tonegawa, G.J. Nossal e Harald von Boehmer dimostrarono che il TCR era fatto in modo tale da legare con forte affinità le molecole HLA non self mantenendo invece una bassissima affinità per quelle self. Il forte legame con l’HLA non self rappresentava il momento di pre-attivazione del linfocita T ma, affinchè l’attivazione diventasse completa, era necessario che altre molecole, i recettori co-stimolatori CD28 e CTLA-4 (oggi noto come CD152 del linfocita T), interagissero con le molecole di membrana CD80 o B7-1 e CD86 o B7-2 della membrana delle APC.
costimolazione

Nel 1997, Joseph G. Altin ed Erica K. Sloan, della Australian National University, Canberra, dimostrarono che anche l’interazione tra la molecola CD45 dell’APC e i recettori del linfocita T era necessaria per attivarlo del tutto. Questa rappresentava la fase di costimolazione indispensabile affinché il linfocita T preattivato non morisse o rimanesse bloccato.
Una volta che i linfociti venivano attivati, proliferavano velocemente e producevano un clone di cellule figlie tutte identiche con lo stesso TCR del linfocita attivato che le aveva generate. I linfociti che possedevano il recettore CD4+ erano in grado di secernere varie citochine come l’IL-12, utili per potenziare l’attività dei linfociti citotossici CD8+ da uno stato di pre-attivazione. I linfociti T che possedevano il recettore CD8+ erano per la maggior parte cellule killer in grado di secernere “perforine”, cioè proteine citolitiche in grado di lisare la membrana delle cellule bersaglio facendole suicidare inducendole a produrre una citochina detta TNF (Tumor Necrosis Factor); questo meccanismo era fondamentale per eliminare cellule estranee, mutate e infettate da batteri e virus.
interazione tra l’antigene e il recettore BCR

Altri studi dimostrarono che i linfociti B a differenza dei linfociti T, riconoscevano l’antigene non per i peptidi estranei della molecola HLA come avveniva per i linfociti T, bensì per la forma tridimensionale che assumeva l’antigene stesso. Tale riconoscimento iniziò ad essere compreso negli anni ’90: il riconoscimento dipendeva dall’interazione tra l’antigene e il recettore BCR (B–Cell Receptor) del linfocita B costituito da una molecola immunoglobulinica monomerica legata alla membrana, di un unico isotipo (IgD, IgM, IgA o IgE) che legava il ligando e da un eterodimero chiamato CD79 (Igά/Igβ) che rappresentava la frazione di trasduzione del segnale.
Da questo momento si comprese che il linfocita B così pre-attivato era una APC in quanto l’antigene veniva accolto nel citoplasma del linfocita B dove veniva digerito in peptidi residui di cui alcuni venivano espressi a livello della membrana cellulare insieme alle molecole HLA di classe II. Un linfocita CD4+ poteva così riconoscere con il suo TCR il peptide estraneo sulla membrana del linfocita B e interagire.
segnali di co-stimolazione

A questo punto il linfocita T poteva liberare citochine come altrettanti segnali di co-stimolazione facendo attivare il linfocita B che poi, proliferando, poteva dar luogo ad un clone piuttosto numeroso di cellule che si potevano differenziare sia in plasmacellule che in cellule B della memoria; le plasmacellule erano a loro volta in grado di produrre una grande quantità di anticorpi.
Negli anni ’90 si assistette inoltre a una svolta radicale dei paradigmi che si riferivano a come il sistema immunitario discriminava ciò che apparteneva o meno all’individuo (self Versus non-self). Particolare attenzione fu prestata ad una popolazione di linfociti, le cellule NK, scoperte verso la fine degli anni ‘70. Fino alla fine degli anni ‘80 esistevano su queste cellule solo descrizioni di tipo morfologico e poco si conosceva su come e perché potessero uccidere cellule tumorali non avendo alcun effetto contro le cellule normali.
RECETTORI SULLE CELLULE NATURAL KILLER

Per lungo tempo non si comprese la loro funzione citotossica contro le cellule tumorali all’interno del sistema immunitario. Agli inizi degli anni’90 si notò che le cellule NK svolgevano la loro azione citotossica su cellule che non possedevano i recettori MHC e nel riconoscimento dell’antigene da parte dei leucociti: su queste osservazioni si formulò l’ipotesi che le cellule NK possedessero recettori del tutto peculiari, capaci di inibirne la funzione quando interagivano con MHC normale. L’immunologo genovese Lorenzo Moretta dimostrò per primo l’esistenza di questi recettori e ne definì la funzione.
Nei primi anni del XXI secolo ai già noti meccanismi di tolleranza periferica, quali l’anergia, il network idiotipico, la morte cellulare per apoptosi indotta dall’attivazione, la segregazione anatomica degli antigeni self, si sono aggiunte le scoperte di varie popolazioni di cellule T regolatorie, definite “Treg” (Regulatory T cell), che giocherebbero un ruolo chiave nella tolleranza periferica.
IMMUNOTERAPIA
Nel 2007 è stato anche identificato un altro subset di cellule T CD4+ denominato “Th17”, fortemente implicato nell’immunità innata (infezioni specie verso batteri extracellulari) oltre che nei fenomeni autoimmuni (sclerosi multipla, LES, artrite reumatoide).
Con la scoperta, a partire dagli anni’70, degli anticorpi monoclonali, lo scenario dell’immunologia è cambiato, trovando sempre nuovo vigore, tanto da rendere più vasto lo spettro dei suoi studi.
Questi hanno permesso la messa a punto di agenti anti-infiammatori (es: anti-TNF), l’immunoterapia nel trattamento di alcuni tumori e lo sviluppo di nuovi farmaci biologici (inibitori del check-point immunitari) ottenendo una maggiore sopravivenza e qualità della vita dei pazienti oncologici. Sono stati individuati nuovi adiuvanti per attivare ed indirizzare meglio le difese più adatte. L’introduzione di terapie cellulari dove è stato possibile “rieducare” e “ingegnerizzare” in vitro cellule effettrici contro cellule leucemiche e linfoidi (CAR – T).
Anche nel campo dei trapianti d’organo le conoscenze acquisite nei diversi settori della fisiopatologia del rigetto di organi e tessuti trapiantati ha visto la nascita di nuovi farmaci anti-rigetto più specifici e selettivi che, seppure ancora a livello sperimentale, hanno contribuito in maniera significativa ad una maggiore sopravvivenza del trapianto, alla diminuzione di dosi elevate dei farmaci immunosoppressivi e al loro cronico utilizzo.
SFIDE DEL FUTURO
Ma le più grandi sfide nel prossimo futuro dell’immunologia saranno quelle rivolte al capitolo :
1. degli xenotrapianti attraverso le tecnologie dell’ editing genomico (CRISPR- Cas , Talen) per permettere una modifica dei geni tanto da poter potenzialmente rivoluzionare l’immunologia dei trapianti non solo per superare le barriere nell’ambito dello xenotrapianto ma anche per le molte patologie a componente genetica;
2. della prevenzione passando da vaccini realizzati da patogeni interi o tossine inattivate chimicamente :
a) a vaccini ricombinanti (mediante la tecnica del DNA ricombinante) ,
b) a vaccini di DNA che si basano sulla possibilità di far produrre alle cellule dell’organismo da immunizzare gli antigeni che devono scatenare la risposta immunitaria. Ciò può avvenire in due modi: infettando le cellule con un virus non citopatico (cioè che non le uccide) oppure inoculando un plasmide contenente un cDNA
c) vaccini a mRNA come quelli prodotti di recente , in risposta alla pandemia di Covid -19 che utilizzano frammenti di mRNA virali nelle cellule umane all’interno di nanoparticelle lipidiche per raggiungere la loro destinazione ancora integri.
L’immunologia è tuttora da considerarsi sia come area di ricerca che come strumento di diagnosi e terapia, una branca della scienza in rapida evoluzione e la sua storia è ben lungi dal considerarsi conclusa.
Bibliografia:
- Miller J.F.A.P. (1961) “The immunological function of the thymus“. Lancet 2 ; 748
- Benacerraf B., McDevitt H.O. (1972) “The histocompatibility linked immune response genes“. Science 175; 273.
- Porter, R.R. (1973). “Structural studies of Immunoglobulins“. Science 180; 713 – 716.
- Edelman, G.M. (1973). “Antibody structure and molecular immunology“. Science 180; 830 – 840.
- Steinman, R.M. and Cohn, Z.A.(1973). “Identification of a novel cell type in peripheral lymphoid organs of mice“. I. Morphology, quantitation, tissue distribution organs of mice. J. Exp. Med. 137; 1142 – 1162.
- Jerne, N.K. (1974). “Towards a network theory of the immune system“. Ann. Immunol. 125C(1-2); 373 – 389.
- Zinkernagel, R.M. and Doherty, P.C. (1974). “Restriction of in vitro T cell-mediated cytotoxicity in lymphocytic choriomeningitis within a syngeneic or semiallogeneic system“. Nature 248; 701 – 702.
- G. Köhler & C. Milstein (1975). “Continuous cultures of fused cells secreting antibody of predefined specificity“. Nature 256 (5517); 495 – 497
- Allison. J.P., McIntyre, B.W., Bloch, D. (1982). “Tumor-specific antigen of murine T-lymphoma defined with monoclonal antibody“. J. Immunol. 129(5); 2293 – 2300.
- Tonegawa, S. (1983). “Somatic generation of antibody diversity“. Nature 302; 575 – 581.
- Marrack, P. and Kappler, J. (1986). “The T cell and its receptor“. Sci. Am. 254(2); 36 – 45.
- Bjorkman, J.B., Saper, M.A., Samraoiu, B., Bennett, W.S., Strominger, J. and Wiley D.C. (1987). “Structure of the human class I histocompatibility antigen, HLA-A2“. Nature 329; 506 – 512.
- Bluestone J.A. & Abbas A.K (2003). “Natural versus adaptive Regulatory T cells“. Nat. Rev. Immunol. 3(3); 253 – 257
- Shimizu J., Yamazaki S., Takahashi T. et al. (2002). “Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance“. Nat Immunol. 3(2); 135 – 142
- Zheng SG, Wang J, Wang P. (2007). “IL-2 is essential for TGF-beta to convert naive CD4+CD25- cells to CD25+Foxp3+ regulatory T cells and for expansion of these cells“. J Immunol. 178; 2018 – 2027.
- Moretta, L., Moretta, A. (2004). “Unravelling natural killer cell function: triggering and inhibitory human NK receptors“. EMBO J. 23: 255-259.