L’evoluzione tecnologica degli approcci di editing genomico e la descrizione del sistema CRISPR/Cas
di Sergio Barocci, docente di Immunologia dei trapianti, Università di Genova per la terza età e divulgatore scientifico per la biochimica.
Introduzione

Metodi per modificare il genoma di cellule e organismi sono stati utilizzati da alcuni ricercatori già da diversi anni, specialmente per effettuare approcci di “reverse genetics”, una strategia che consiste nel comprendere le funzioni di un gene analizzando gli effetti che una sua alterazione, o eventualmente inattivazione, provoca in cellule o in interi organismi.
La reverse genetics si contrappone alla strategia opposta chiamata “forward genetics” , nella quale si parte, invece, dalla valutazione di un fenotipo alterato (tipicamente un quadro patologico) cercando di risalire alle sue basi genetiche, identificandone il gene responsabile (Fig.1).
Queste procedure, utilizzate per effettuare l’editing genomico, erano applicabili o a modelli cellulari molto semplici come ad esempio i lieviti che avevano, però, una limitata rilevanza per lo studio delle malattie umane oppure applicabili a organismi più vicini all’uomo, come il “gene targeting“ nel topo, una tecnica biologica per modificare i geni di grande aiuto nel campo della ricerca biomedica.
Gene targeting
|
|
|
|



Il gene targeting è una procedura che si serve della ricombinazione omologa (processo ubiquitario che avviene grazie all’azione di ricombinasi endogene presenti nelle cellule) per modificare un gene. Si tratta di una metodica, messa a punto alla fine degli anni ’80 del secolo scorso da Mario Capecchi (Fig. 2) , insieme a Martin Evans (Fig. 3) e Oliver Smithies (Fig. 4) allo scopo di permettere la cancellazione di un gene, la rimozione di esoni o l’introduzione di mutazioni puntiformi e che ha valso loro il Premio Nobel per la Medicina nel 2007.
Le ricombinasi sono enzimi in grado di coordinare una ricombinazione genetica , ossia uno scambio di regioni di DNA a partire da segmenti distinti e separati tra loro.
Le ricombinasi più note per attività biologica ed utilizzo nelle tecniche di ingegneria genetica sono rappresentate da :
a) Cre ricombinasi, topoisomerasi di tipo I proveniente dal batteriofago P1, molto utilizzata in applicazioni di biologia molecolare;
b) Flp ricombinasi, usata anch’essa nelle ricombinazioni in vitro;
c) RecA, responsabile delle ricombinazioni in Escherichia coli;
d) Hin ricombinasi, responsabile delle ricombinazioni in Salmonella;
e) RAD51, responsabile delle ricombinazioni in diversi organismi eucarioti.
Il gene targeting (Fig. 5 ) in breve, consiste nel creare filamenti di DNA con dei batteri (formati dal gene target che può essere mutato o deleto affiancato da una cassetta di selezione positiva come ad esempio la cassetta Neo per la resistenza alla geneticina, un antibiotico aminoglicosidico simile alla gentamicina, composto tossico per le cellule).
DNA ARTIFICIALE

Successivamente in base al gene che si desidera inattivare, vengono affiancate altre sequenze omologhe. Sono presenti anche sequenze omologhe negative.
In breve , il “DNA artificiale” viene inserito dentro le cellule staminali embrionali di un topo per essere poi prese da una blastocisti (una fase embrionale che rappresenta la diretta evoluzione dell’ embrione) appartenente ad un topo. Queste cellule vengono irradiate. Le cellule embrionali che danno i migliori risultati vengono immesse in una blastocisti che sarà poi, a sua volta, inserita dentro un topo femmina in stato di gravidanza.
Si dà vita, così, a topi speciali, detti topi chimera cioè aventi più di un tipo di cellule geneticamente diverse , sempre che le cellule genitali si inseriscano bene nella cresta urogenitale. I topi chimera, successivamente, vengono fatti incrociare con dei topi wild type per ottenere gli eterozigoti.
Uso delle nucleasi nell’editing genomico

Un notevole avanzamento nelle procedure di editing genomico si è avuto quando è stato introdotto l’utilizzo delle “nucleasi”, cioè proteine enzimatiche in grado di rompere il doppio filamento del DNA in punti specifici (le cosiddette “forbici molecolari” ).
Ma per quale motivo le nucleasi sono in grado di attivare un processo di editing genomico ? Semplicemente perché la rottura del DNA non può essere tollerata dalla cellula, la quale immediatamente attiva un processo di riparazione che può avvenire mediante due seguenti modalità:
- Non homologous end-joining (NHEJ) cioè unione delle estremità non omologhe.
Questa procedura agisce sia rapidamente che efficacemente e non si avvale di una molecola di DNA che funge da stampo. La saldatura delle estremità non-omologhe ha alte probabilità di introdurre differenze ed errori rispetto alla sequenza del DNA prima della rottura e qualora la rottura si verificasse all’interno di un gene che produce una proteina, un probabile effetto di questo processo sarebbe la mutazione del gene e la possibile inattivazione della proteina corrispondente, con importanti applicazioni nel campo della reverse genetics (Fig. 6) - Homologous recombination (HR) o ricombinazione omologa. Questa modalità di riparazione del DNA richiede la presenza di una molecola di DNA stampo identica (o quasi) a quella pre-rottura che può essere anche fornita esternamente. Risulta meno efficiente rispetto alla saldatura non-omologa, però è in grado di riparare il DNA ricopiando perfettamente, senza alcun errore, la sequenza della molecola fornita come stampo. È proprio quest’ultima modalità che è in grado di fornire le maggiori prospettive per l’utilizzo dell’editing genomico a scopi terapeutici (Fig. 6).
tipi diversi di nucleasi

Nei primi anni del 2000 per poter modificare il genoma di organismi modello vennero sono stati introdotti sistemi di editing genomico basati su tipi diversi di nucleasi, tra cui le meganucleasi, le nucleasi a dita di zinco (Zinc finger o ZFN), le nucleasi che mimano gli effettori dell’attivazione transcrizionale (TALEN o Transcription activator-like effector nucleases) (Fig. 7 ) e le nucleasi guidate da RNA come la Cas 9, associata a brevi ripetizioni palindrome raggruppate e separate ad intervalli regolari (clustered regularly interspaced short palindromic repeats o CRISPR).
Le meganucleasi, scoperte verso la fine degli anni ’80 del secolo scorso, sono enzimi della famiglia delle endonucleasi comunemente presenti nei batteri e capaci di riconoscere e tagliare sequenze molto ampie di DNA. Le meganucleasi più diffuse e conosciute sono le proteine della famiglia LAGLIDADG che devono il loro nome ad una sequenza di aminoacidi conservata. Sebbene ne esistano, in natura, diverse forme (ognuna dotata di piccole e utili differenze relative alle zone del DNA che possono tagliare), la probabilità che siano adattabili alle esigenze specifiche dell’ ingegneria genetica è virtualmente nulla ed è proprio per questo motivo che prima di essere usate devono essere sottoposte a un processo di manipolazione. Pur avendo le meganucleasi un grado di tossicità cellulare inferiore rispetto alle nucleasi delle dita di Zinco o ZFN , il lavoro di adattamento necessario per il compito che andranno a svolgere risulta più costoso e richiede più tempo.
ZINC FINGER

I Zinc finger o letteralmente “dita di zinco” (ZFN) sono delle nucleasi o meglio delle regioni proteiche capaci di agganciarsi al DNA, caratterizzate dalla presenza di un atomo di zinco. Sono composte da un dominio di taglio del DNA (costituito da due unità dell’enzima di restrizione FokI di tipo IIS) ) e un dominio di legame al DNA, a sua volta formato da moduli con struttura zinc-finger.
Ogni modulo può legare tre o quattro nucleotidi adiacenti e il legame avviene attraverso alcuni residui aminoacidici ‘chiave’, che entrano direttamente in contatto con il solco maggiore del DNA. Sostituendo tali aminoacidi è possibile modificare la specificità del singolo modulo. Sono stati creati dei moduli sintetici che riconoscono quasi tutte le 64 possibili triplette nucleotidiche, e che possono essere fusi in modo da riconoscere sequenze specifiche, quelle che si vogliono modificare. I moduli sintetici riconoscono il locus che interessa (sulla base della sua sequenza di DNA) e i due monomeri FokI lo tagliano. Nella pratica, la creazione del dominio di riconoscimento è un processo lungo e laborioso, in quanto ogni modulo mantiene le sue caratteristiche di legame solo nel contesto dei moduli adiacenti. Ciò significa che la specificità di un modulo può cambiare a seconda dei moduli che ad esso sono fusi e questo fenomeno può portare alla formazione di nucleasi non specifiche per la sequenza desiderata, ma che possono tagliare il genoma anche in sequenze diverse (Fig. 8).
TALEN

Anche le TALEN sono nucleasi basate sull’associazione fra due unità di nucleasi FokI e proteine che riconoscono e si legano in modo selettivo alle sequenze nucleotidiche prescelte. Tali proteine sono state prodotte partendo da fattori di trascrizione presenti in batteri del genere Xanthomonas, detti transcription activator-like effectors (TALE).
Anche in questo sistema, il dominio di riconoscimento del DNA è modulare composto da ripetizioni di 33-35 aminoacidi e ogni ripetizione è in grado di riconoscere un singolo nucleotide nella sequenza di DNA target. Pertanto, esistono soltanto quattro diversi moduli TALE capaci di riconoscere i rispettivi quattro nucleotidi del DNA. Così come per le ZFNs, anche i singoli moduli TALE possono essere legati in serie per formare domini di legame al DNA, in grado di riconoscere la sequenza di DNA di interesse, ed inoltre domini di taglio di FokI . A differenza dei moduli a dita di zinco, l’interazione con il DNA dei singoli moduli TALE non è ostacolata dai domini vicini e quindi generare nuove nucleasi artificiali basate su struttura TALE (TALENs) con diverse specificità è semplice e veloce e può essere fatto in ogni laboratorio, utilizzando tecniche di clonaggio standard (Fig. 9)
Tutti questi sistemi presentano un’ottima efficacia ma hanno lo svantaggio di richiedere la produzione di proteine specifiche per ogni sequenza che si voglia tagliare e quindi comportano costi elevati che non sono alla portata di laboratori medio – piccoli.
Il sistema Crispr/Cas9 ha dimostrato, fin da subito, una potenzialità e una versatilità fino a poco tempo prima inimmaginabili: qualunque tipo di cellula vegetale, animale, inclusa quella umana, può essere modificata geneticamente e la correzione può avvenire anche per un singolo e minimo errore, e ovunque nel genoma. Inoltre, è una tecnica facile da utilizzare, veloce ed economica, il che ne amplia le potenzialità, specialmente. in ambito terapeutico.
Cosa è cambiato con l’avvento della tecnologia CRISP-Cas9 e cosa sta cambiando?

CRISPR è una sorta di semplice ma ingegnoso sistema immunitario che i batteri utilizzano per difendersi dai virus (Fig. 10a). È costituito da una serie di sequenze genomiche ripetute intervallate a frammenti di sequenze virali.
Questa regione produce dei piccoli RNA non codificanti per proteine denominati crRNA. Se la cellula batterica viene infettata da un virus la cui sequenza genomica è riconosciuta da uno di questi crRNA non codificanti si avrà la formazione di una molecola ibrida crRNA-DNA virale. La parte ripetuta del crRNA ha un segnale di riconoscimento per una nucleasi Cas (abbreviazione per CRISPR – associated), così denominata perché il gene corrispondente è adiacente alla regione CRISPR nel genoma batterico, che taglia il DNA virale e pertanto neutralizza l’infezione. L’intero sistema ha preso il nome di CRISPR/Cas.
Esso è attualmente il sistema più promettente per semplicità d’uso e per specificità. A differenza di ZFN e TALEN, nel sistema CRISPR/Cas9 il riconoscimento della sequenza di DNA da modificare non è operata tramite la nucleasi ma da una sequenza di RNA. Le sequenze di RNA sono oggetto di più facile manipolazione genica rispetto alle proteine (nucleasi) e questa caratteristica ha permesso di aprire notevoli prospettive alle tecniche di editing genomico. In particolare, il sistema CRISPR – Cas 9 è stato adattato in modo da sostituire, alle sequenze virali contenute nei cr RNA, degli RNA sintetici di 18 – 20 basi , denominati RNA guida o sgRNA corrispondenti ad una qualsiasi sequenza genomica bersaglio specifica.
cas9
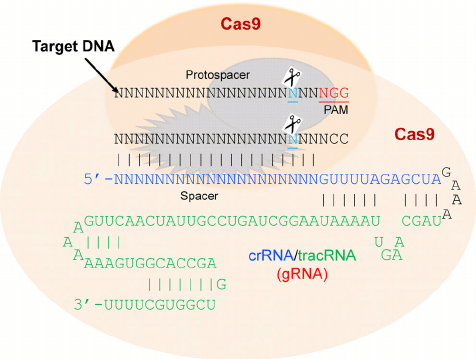
Come nucleasi viene utilizzata la Cas9, isolata in Streptococcus pyogenes, molto maneggevole a causa delle sue ridotte dimensioni. Questo sistema è stato scoperto negli anni ‘80 del secolo scorso nell’Escherichia coli e poi caratterizzato come un sistema immunitario adattativo in molti batteri e archeobatteri. Quando il DNA estraneo entra nel batterio, esso viene degradato dalla nucleasi Cas9.
La specificità rispetto al DNA bersaglio è legata, come già espresso in precedenza, ad una piccola di molecola di 18 – 20 nucleotidi di RNA guida, il cosiddetto CRISPR-RNA (crRNA), codificato nel locus CRISPR e complementare al DNA estraneo. Per il corretto riconoscimento del sito bersaglio è necessaria, anche, una breve sequenza aggiuntiva (di tre nucleotidi, 5’-NGG-3’) adiacente alla sequenza bersaglio, chiamata PAM (Protospacer Adjacent Motif). Un secondo corto RNA, il tracrRNA (trans-activating CRISPR-RNA), si lega al crRNA, e forma un complesso stabile con la proteina Cas9, che presenta due domini nucleasici. Nell’editing genomico, il crRNA e il tracrRNA sono, in genere, fusi insieme per formare un singolo RNA-guida (sgRNA) (Fig. 10b).
Nel 2013 è stato dimostrato che Cas9 di Streptococcus pyogenes può essere utilizzato per il genome editing di cellule umane in coltura, quindi anche in sistemi eucariotici. Da allora, il sistema CRISPR/Cas è diventato il sistema di maggior successo grazie alla facilità di costruire nucleasi con specificità di sequenza e per il fatto che funziona in quasi ogni organismo. Nell’editing genomico, la molecola di sgRNA viene infatti sintetizzata sulla base della sequenza bersaglio e inserita nella cellula insieme al gene che codifica Cas9 o all’enzima stesso.
La storia di CRISPR dalle origini

Come molte grandi scoperte anche quella di CRISPR è avvenuta quasi per caso. La storia di CRISPR inizia nel 1987, con il biologo giapponese Yoshizumi Ishino (Fig. 11) dell’Università di Osaka mentre stava studiando il gene responsabile della conversione isoenzimatica della fosfatasi alcalina, nel comune batterio intestinale Escherichia coli. Durante il sequenziamento di una parte del cromosoma di questo batterio, Ishino scopre diverse brevi sequenze ripetitive di DNA, separate da brevi sequenze spaziatrici casuali (spacer). A quel tempo, era quasi impossibile prevedere la funzione biologica di queste insolite sequenze ripetute (repeat unit) a causa della mancanza di dati sufficienti sulla sequenza del DNA, specialmente per gli elementi genetici mobili. La funzione effettiva di questa sequenza unica è rimasta enigmatica fino ai primi anni 2000.
Origine del termine CRISPR e la prima ipotesi ad opera di F. Mojica

Nel 1993 in Spagna, il microbiologo spagnolo Francisco Mojica (Fig.12), dell’Università di Alicante, lavorando su un procariote (organismo unicellulare), isolato nelle paludi salate di Santa Pola (punto di partenza della storia), con estrema tolleranza alle alte concentrazioni di sale, nota una struttura curiosa nel DNA di quell’organismo: più copie di sequenze ripetitive (20 – 40 nucleotidi) sempre identiche tra loro mentre le sequenze spaziatrici appaiono, invece, sempre diverse e uniche.
Proseguendo con la ricerca, trova queste sequenze particolari anche in altri procarioti (archeobatteri) che le nomina Short Regularly Spaced Repeats o SRSR e con l’acronimo CRISPR (Clustered Regularly Interspaced Palindromic Repeats) nel 2001 insieme al biochimico olandese Ruud Jansen dell’Università di Ultrech.
Scoperta dei geni Cas
Un passo in avanti verso una migliore comprensione della funzione dei CRISPR avviene nel 2002 grazie ancora al contributo di Jansen che osserva che nei procarioti l’insieme (cluster) di ripetizioni è accompagnato da un set di geni omologhi che aiutano a costituire i “sistemi associati a CRISPR” (CRISPR Associated System o geni Cas).
All’inizio vengono scoperti quattro geni Cas ( Cas1, Cas2,Cas3 e Cas4) e solo in seguito si riuscì a caratterizzare la natura dei trascritti di questi geni: proteine che contengono dei domini elicasici e nucleasici e che suggeriscono un ruolo nell’organizzazione tridimensionale dei loci dei CRISPR.
Ipotesi sulla funzione di CRISPR
Nel corso dei primi anni 2000, Mojica dopo aver analizzato questi tratti di DNA in molti batteri, si accorge che le sequenze spaziatrici erano, in realtà, identiche alle sequenze del DNA dei virus batteriofagi che, in precedenza, avevano infettato le cellule batteriche. Era come se i batteri stavano immagazzinando tra le ripetizioni CRISPR, frammenti di DNA di vari ceppi virali. Egli, allora, ipotizza che il sistema CRISPR rappresenti un qualche tipo di difesa batterica contro le infezioni virali. Questa particolare scoperta di Mojica viene grossolanamente sottovalutata in quel momento e solo a partire dal 2005 si arriva ad una ipotesi che si rivela poi corretta nella maggior parte degli Archei e in una grande percentuale di batteri diversi dall’Escherichia Coli.
Infatti, tre gruppi mostrano indipendentemente che alcune di queste sequenze distanziali hanno un’origine dal batteriofago. Nel 2006, Eugene V. Koonin e coll. Suggeriscono che il sistema CRISPR-Cas rappresenta un sistema immunitario basato sull’interferenza dell’RNA procariotico (RNAi) dove il locus CRISPR è in grado riconoscere gli attacchi da virus esogeni mediante la seguente strategia:
• acquisizione di una sequenza spacer, dovuta a una invasione precedente del virus, nel locus CRISPR del genoma batterico;
• trascrizione del DNA-spacer in RNA;
• utilizzo dell’RNA-spacer per riconoscere la nuova invasione da parte dello stesso virus.
La prova arriva più tardi, nel 2007, grazie a due microbiologi statunitensi Rudolphe Barrangou e Philippe Horvath, studiosi dei batteri dello yogurt, che li incubano in presenza di uno specifico batteriofago. Sequenziano, quindi, i genomi dei batteri sopravvissuti e scoprono che le cellule incorporano porzioni del genoma del batteriofago nella regione CRISPR del loro cromosoma. CRISPR, pertanto, rappresenta chiaramente una qualche forma di immunità adattativa contro i batteriofagi: l’infezione porta alla formazione in modo polarizzato di nuove sequenze distanziali e alla protezione contro ulteriori infezioni, che è abolita con la cancellazione delle sequenze distanziali o di alcuni dei geni Cas.
In poche parole, i batteri utilizzano il sistema CRISPR per difendersi dai virus che li attaccano. Dopo una infezione, l’organismo utilizza l’enzima Cas9 per tagliare il DNA del virus, che riconosce grazie a una molecola-guida formata da RNA. Il taglio inattiva il virus e un frammento di quest’ultimo viene immagazzinato nel DNA del procariote, in modo da riconoscerlo immediatamente in caso di nuova infezione. Questa collezione di frammenti di virus è l’equivalente batterico del nostro sistema immunitario. La scoperta di questo meccanismo di difesa segna l’inizio della rivoluzione CRISPR.
Dalle ipotesi di Mojica alle prime scoperte definitive
Dopo la scoperta della funzione di CRISPR nei batteri , si rendono necessari altri cinque anni di ricerca per arrivare alle prime scoperte definitive. CRISPR, cattura l’interesse anche della biochimica statunitense Jennifer Doudna (Fig.13) dell’Università di Berkeley in California. Essa, nutre il sospetto che alcuni geni vicino alla regione CRISPR possano svolgere un qualche ruolo. Infatti, il suo laboratorio è in grado di comprendere il ruolo di due delle proteine enzimatiche prodotte dai geni Cas ma quello che ancora manca è la spiegazione di come funzioni l’intero sistema, cioè come queste proteine Cas (nucleasi) utilizzano l’informazione contenuta tra le sequenza CRISPR come guida, una specie di identikit che servirebbe per rintracciare i virus invasori e distruggerli tagliandone il DNA.

La svolta avviene nel 2011, a seguito di un incontro casuale tra la Doudna e la biologa francese Emmanuelle Charpentier (Fig.13) che nel frattempo studia un altro enzima il Cas 9, presso il Laboratorio di Medicina Molecolare dell’Università Svedese di Umea, individuato nel batterio dello Streptococcus pyogenes. Si tratta di un’endonucleasi con la capacità di tagliare il DNA come un paio di forbici. Per alcuni aspetti, è simile agli enzimi di restrizione scoperti decenni prima. Gli enzimi di restrizione hanno la funzione di tagliare il DNA in una breve sequenza specifica. L’enzima EcoRI, ad esempio, taglia sempre a livello della sequenza GAATTC. Cas9, invece, è un enzima programmabile che può tagliare il DNA in una qualsiasi sequenza di nucleotidi se gli si dice dove farlo. Le istruzioni per il taglio provengono da una piccola molecola di RNA che si lega all’enzima Cas9 per formare un complesso ribonucleoproteico :
1. Un crRNA (CRISPR-RNA):
2. Un trascrRNA (trans-activating CRISPR RNA).
Sia la Doudna che la Charpentier riescono a mettere insieme tutti i pezzi del puzzle, comprendendo il modo (come un sistema immunitario adattativo) con cui opera il sistema CRISPR nei batteri contro i virus.
Quando un virus infetta i batteri, per prima cosa inietta il suo DNA nella cellula, il DNA virale poi istruisce i batteri su come far creare copie del virus. La cellula segue le istruzioni, produce centinaia di nuovi virus e alla fine si distrugge, consentendo ai nuovi virus di uscire. Tuttavia, se per qualche motivo un batterio riesce a sopravvivere all’infezione e non si distrugge, prende un frammento del DNA virale e lo unisce al proprio cromosoma. Dopodiché aggiunge sequenze di ripetizione CRISPR su entrambi i lati del DNA virale. In questo modo, il batterio crea una sorta di database di virus conosciuti, Questo elenco viene trasmesso geneticamente a tutte le cellule discendenti in modo che le generazioni successive siano in grado di riconoscere i virus ancora prima di venire attaccate.
Una volta che il batterio ha acquisito la sequenza di DNA spaziatore nel locus CRISPR, quando lo stesso tipo di virus lo infetta nuovamente, il DNA spaziatore della regione CRISPR è trascritto in RNA che, legandosi all’enzima Cas9, grazie alla complementarietà delle basi, è utilizzato dalla cellula per riconoscere il DNA virale dei nuovi invasori.
In realtà, quando i batteri sono infettati trascrivono l’intera regione CRISPR in RNA, che poi viene scisso in pezzi separati detti crRNA. Ogni crRNA, così ottenuto, contiene una ripetizione CRISPR con un breve tratto di 20 nucleotidi di DNA virale. Il crRNA è quindi caricato nell’enzima Cas9, ancorandolo a un altro pezzo di RNA chiamato tracrRNA.
tracrrna

Charpentier ha già scoperto il tracrRNA insieme a Cas9, ma non è in grado di determinarne la funzione. Ora, invece, si comprende che tracrRNA funziona da sito di collegamento universale per il crisprRNA. Quindi, l’enzima Cas9 può essere programmato dalla cellula per tagliare a livello di qualsiasi sequenza target sia specificata alla fine del crisprRNA (Fig.14).
Se il DNA virale entra nuovamente nella cellula, il complesso CRISPR-Cas9 riconosce il DNA mediante l’accoppiamento di basi complementari, lo apre e lo taglia. Una volta tagliato, il DNA virale non è più in grado di danneggiare la cellula.
CRISPR-Cas9 è, pertanto, un sistema straordinario con la capacità di apprendere e adattarsi. Non è molto diverso dal nostro sistema immunitario adattativo, che impara a riconoscere i microrganismi nocivi e produce anticorpi altamente specifici per combatterli. Una volta compreso il meccanismo CRISPR-Cas9, la Doudna si domanda se sia possibile personalizzare il sistema CRISPR creando il proprio crRNA e caricandolo nell’enzima Cas9.
Doudna, Charpentier e Šikšnys

Allora, la Doudna e la Charpentier ri-ingegnerizzano l’endonucleasi Cas9 in un sistema a due componenti molto più maneggevole fondendo le due molecole di RNA in un unico RNA denominato single-guide RNA che, quando fuso a Cas9, è in grado di cercare e tagliare il DNA-target specificato da questo. Manipolando la sequenza del single-guide RNA, il sistema artificiale Cas9 può essere ingegnerizzato in maniera tale da riconoscere e tagliare qualsiasi sequenza di DNA.
Si arriva così alla descrizione puntuale di questa tecnica di editing che è pubblicata nel 2012 sulla Rivista Science. Per questo lavoro, entrambe, sono insignite nel 2020 del Premio Nobel per la Chimica per lo sviluppo di un metodo di editing genomico basato su CRISPR.
DOUDNA, CHURCH E ZANG

Da notare che anche il biochimico lituano Virginijus Šikšnys (Fig. 15), presso l’Istituto di Biotecnologia dell’Università di Vilnius, elabora contemporaneamente alla due ricercatrici il funzionamento di CRISPR-Cas9, ma la pubblicazione della sua ricerca viene ritardata da un’approfondita peer review da far così trascurare il suo lavoro.
Pochi mesi dopo, dalla pubblicazione su Science, nel 2013, seguono tre importanti studi che dimostrano la possibilità di utilizzare questa innovativa tecnica per modificare il genoma in maniera mirata sulle cellule umane (in coltura, in vitro): uno a firma di J. Doudna, un altro dal genetista statunitense George Church (Fig. 16) dell’Università di Harvard e l’ultimo dal biochimico cinese Feng Zhang (Fig. 17) del Broad Institute del MIT . Questi studi, risultano le pubblicazioni pioniere su CRISPR.
UN INTERESSE SEMPRE MAGGIORE

Da allora, un certo numero di studi sul tema pubblicati nelle maggiori riviste internazionali incominciano ad avere una crescita esponenziale, un ritmo tale che ancora oggi non accenna a diminuire. CRISPR dimostra fin da subito una potenzialità e una versatilità fino a poco prima immaginabile. Qualunque tipo di cellula animale, vegetale ed umana può essere modificata geneticamente in maniera programmata e la correzione può avvenire anche per un singolo errore e ovunque nel genoma.
Il sistema CRISPR, si basa quindi sulla combinazione di due elementi: un enzima Cas ed un RNA guida che si appaia al DNA per indicare a Cas il punto in cui tagliare. Come nel caso della terapia genica, anche la strategia di editing basata su CRISPR può essere somministrata in vivo cioè direttamente nell’organismo o ex vivo cioè all’esterno su cellule vive prelevate dall’organismo. Sono molte le applicazioni CRISPR.
L’impatto maggiore l’ha avuto, però, in ambito biomedico spaziando dalle malattie genetiche, specialmente quelle rare come la distrofia muscolare di Duchenne, la beta -talassemia e la fibrosi cistica, alle neoplasie del sangue (leucemia, linfoma e mieloma multiplo) passando per le malattie neurologiche (malattia di Alzheimer e malattia di Parkinson) fino alle malattie infettive come l’infezione da HIV per la quale l’inattivazione del gene codificante per la proteina CCR5 sembra avere un effetto terapeutico. Questa proteina, svolge un ruolo essenziale per l’ingresso del virus nella cellula e quindi la sua completa inattivazione mediante l’editing genomico potrebbe dare risultati promettenti nei confronti dell’infezione da HIV.
Inoltre, l’utilizzo della tecnologia CRISPR è anche studio nel campo degli xenotrapianti, in particolar modo degli organi dei suini, per la terapia di patologie epatiche, cardiache e renali.
Finora, il limite per l’utilizzo degli xenotrapianti è rappresentato dal fatto che questi animali possono essere infettati da retrovirus (Perv) che si installano nel DNA delle cellule. Oggi, è possibile dimostrare come la tecnica CRISPR sia in grado di distruggere, nei maiali, almeno una sessantina di aree del genoma dove si sono insediati questi virus dannosi.
Come accade spesso per tutti i farmaci sperimentali, occorre molta cautela prima di poter parlare di successo e soprattutto di cura. I benefici devono durare nel tempo e devono essere dimostrati con un certo numero di pazienti. Tuttavia, le storie dei primi pazienti trattati con CRISPR fanno oggi ben sperare anche se la strada è ancora lunga.
Infatti, il processo di CRISPR- Cas9 può portare spesso ad errori di vario tipo che rendono il gene inattivo, in quanto la classica Cas 9 può svolgere solo l’attività di tagliare un gene e di inattivarlo.
Una funzione importante ma non sufficiente specialmente per affrontare malattie genetiche complesse e il suo impiego rimane, pertanto, limitato. Se l’obiettivo è quello di sostituire una sequenza, bisogna fornire alla cellula anche una copia corretta del gene che sia complementare alle estremità del sito di taglio e che la cellula utilizzerà come stampo per la correzione dopo il taglio effettuato dal sistema di editing.
In aggiunta, questa tecnica di editing può anche causare problemi di tossicità e di oncogenicità che possono essere ridotti se le modifiche vengono fatte chimicamente come avviene nel base editing.
Nel 2015 si descrive un’alternativa al sistema CRISPR- Cas9 , il sistema CRISPR / Cpf1 dal batterio Francisella novicida o CRISPR- Cas12a, più economico e in grado di cambiare strutture più complesse.
Applicazioni di modifica del genoma : sistemi di editing genomico verso la riduzione degli errori e delle imprecisioni
Base editing

A partire dal 2016, la tecnologia CRISPR si evolve ancora: non è più un sistema basato sul meccanismo di taglia – incolla ma un correttore che non necessita di forbici molecolari.
Grazie alla biologia molecolare, si iniziano a confezionare varianti di CRISPR, aggiungendo degli effettori cioè delle parti in grado di affinare e ampliare le capacità del bisturi molecolare. Sono nati così gli editor base capaci di apportare modifiche specifiche alla base del DNA che consistono in una Cas alterata (dCas9 o Cas nickase) fusa con una deaminasi, o di sostituire una singola base (CBEs o editor di basi di Citosina: cambi di base da C – G ad A – T e ABEs o editor di basi di adenina : cambi di base da A – T a C – G ) o ancora un intero gene (PEs).
Nel 2017, due studi, uno pubblicato su Nature dal biologo statunitense David Liu (Fig. 18) del Centro di Ricerca Biomedica e genomica (Broad Institute del MIT) e dell’Università di Harvard e l’altro su Science da Zhang presentano delle novità in questo ambito.
Il primo studio dimostra che è possibile correggere la sequenza bersaglio senza tagliare la doppia elica, escludendo le imprecisioni dovute ai meccanismi automatici di riparazione cellulare e riducendo al minimo gli errori. In questo caso CRISPR identifica una singola base errata in un punto specifico del DNA e la modifica chimicamente.
EDITOR BASE

A differenza degli strumenti di editing come CRISPR-Cas9 e Zinc finger che si basano sul taglio del DNA, già entrati da alcuni anni nelle sperimentazioni cliniche, gli editor base, o ‘base editor‘, agiscono in maniera più sofisticata e potenzialmente più sicura. Questi combinano la precisione del sistema CRISPR-Cas9 con le deaminasi, particolari enzimi che possono modificare il genoma cambiando una base azotata in un’altra. Le modifiche fattibili riguardano la base azotata Citosina nella base azotata Timina (C in T) o la base azotata Adenina nella base azotata Guanina (A in G) che sono in grado di coprire oltre il 60% delle mutazioni patologiche. Questi editor base sono rappresentati da tre componenti: un RNA guida (gRNA) che conduce il sistema di editing a termine, l’enzima Cas9 modificato che intacca il DNA sul filamento opposto alla base azotata bersaglio e l’enzima deaminasi che trasporta una base azotata in un’altra. A completamento della modifica e all’adesione del filamento all’altro dopo l’intervento dell’editing, giungono i meccanismi di riparazione cellulare (Fig.19). In questo modo, diventa possibile convertire le lettere che compongono l’informazione genica, sia in cellule umane che nei batteri. Rimane, però, il fatto che questo meccanismo non è in grado di effettuare tutte le conversioni possibili e, di conseguenza, la ricerca continua.
cas13

Zhang autore, invece, dello studio pubblicato su Science, sperimenta una nuova variante Cas chiamata Cas13 (tipo VI), capace di modificare a livello chimico una lettera dell’RNA messaggero o mRNA (la molecola che veicola le istruzioni contenute nel DNA sino al prodotto finale funzionante, la proteina) e non del DNA (Fig.20). La novità di tale studio sta nel fatto che agendo sull’RNA e non sul DNA, tutte le azioni di editing diventano reversibili. Entrambe le varianti Cas9 e Cas13 sono state studiate in modelli cellulari di patologie umane, tra cui l’anemia di Fanconi (un difetto ereditario della riparazione del DNA causato da mutazioni), l’anemia falciforme (malattia ereditaria la cui caratteristica patologica è la presenza di una variante patologica dell’emoglobina, l’emoglobina S o HbS che prende origine da una mutazione puntiforme sul gene della beta-globina che porta a gravi conseguenze come la forma a falce dei globuli rossi e alla loro rigidità e come conseguenza la formazione di trombi, rischio di ictus e anemia emolitica) e l’emocromatosi (altra malattia caratterizzata da un eccessivo accumulo di ferro).
Prime editing

Dal 2017 in poi, diversi gruppi di ricerca nel mondo cercano di creare editor più efficienti e precisi ampliando le possibilità terapeutiche per aumentare, diminuire o sopprimere del tutto l’attività di un gene, sino al prime editing, una nuova tecnica che vede imprecisioni ed errori ulteriormente ridotti, cioè capace di correggere direttamente un gene errato riscrivendone una copia e di commettere solo pochi errori.
Il prime editing è la tecnologia di editing genomico più innovativa e maggiormente applicabile descritta nel 2019 da Liu sulla Rivista Nature, che diventa in poco tempo la tecnica di editing più utilizzata. E’ in grado di apportare un cambiamento al genoma senza inserire modifiche indesiderate al di fuori del bersaglio, i temuti “off target“, in poche parole, una ricerca e una sostituzione dell’editing del genoma senza interruzioni del doppio filamento o del DNA del donatore.
L’RNA guida non solo indica dove si deve effettuare la modifica ma suggerisce anche quali sono gli errori che si devono correggere. In questo preciso caso, non vi sono né tagli della doppia elica del DNA, né modifiche a livello chimico.
Il sistema CRISPR è fuso con una trascrittasi inversa, che utilizza gRNA come stampo e lo ricopia poi sotto forma di DNA, correggendo la mutazione del gene di interesse in modo più preciso. Poiché, non vi è il doppio taglio, si riduce la possibilità di mutazioni “off-target” e dato che si utilizza uno stampo creato su misura, la correzione risulta alla fine più precisa e malleabile.
Per valutarne le sue capacità, il prime editing è stato testato in vitro su modelli cellulari di alcune patologie diverse dal punto di vista della natura dell’errore sulla molecola di DNA, in particolar modo sull’anemia falciforme in cui vi è una base azotata errata (una adenina rimpiazza una timina provocando la sostituzione dell’aminoacido acido glutammico con un altro aminoacido, la valina), sulla malattia di Tay – Sachs (malattia rara causata da un deficit dell’enzima esosaminidasi A che provoca un accumulo del ganglioside GM2 nel cervello) e sulla fibrosi cistica (causata da mutazioni nel gene CFTR che codifica una proteina che controlla il passaggio di acqua e di alcuni sali all’interno e all’esterno delle cellule; la proteina mutata non funziona in modo appropriato e porta alla produzione di muco denso e sudore molto ricco di sali).
Al di là di possibili applicazioni cliniche, oggi ancora lontane, il prime editing rappresenta un importante strumento per la ricerca di base per tutta una serie di patologie in grado di correggere all’incirca l’89% delle mutazioni causanti le malattie genetiche umane (Fig.21).
nanoparticelle lipidiche

Una frontiera promettente nell’editing genomico è rappresentata dalle nanoparticelle lipidiche che si prestano bene a trasportare gli ingredienti chiave del sistema genetico CRISPR sotto forma di RNA (Fig. 22). Infatti, in queste goccioline possono essere inserite le istruzioni per Cas9 (un mRNA che codifica per Cas 9 necessario per il taglio di precisione) o addirittura un base editor (quella versione aggiornata di CRISPR capace di correggere il DNA senza recidere la doppia elica) e l’RNA guida specifico per il gene che causa la patologia. Tutto questo è direttamente iniettato nell’organismo del paziente. Questo approccio è stato usato per il trattamento in vivo dell’amiloidosi da accumulo di transtiretina (TTR), una malattia neurologica rara causata dalla forma difettosa di una proteina prodotta nel fegato.
La strategia da adottare, consiste nel mettere knockout il gene che codifica TTR, lasciando i sistemi naturali della cellula a rinsaldare la lesione in maniera casuale.
Un altro trial clinico in corso è quello per i pazienti affetti da ipercolesterolemia familiare eterozigote [un sottotipo comune e potenzialmente pericoloso per la vita della malattia cardiovascolare aterosclerotica (ASCVD)], laddove ad essere colpito è il gene PCSK9, ma in questo caso il sistema CRISPR utilizzato è un editor base con le nanoparticelle lipidiche che hanno il compito di portare la Cas9 e la sua RNA guida a destinazione nelle cellule epatiche, per assemblare una proteina modificatrice di basi, che poi agendo sul gene PCSK9 di quelle cellule introduce il piccolo errore, trasformando una adenina in una guanina che lo disattiva.
Perchè queste nanoparticelle lipidiche possano essere indirizzarle in maniera preferenziale verso i distretti corporei, il lavoro da fare è ancora molto. Quello che attualmente si sta provando è di individuare diverse formulazioni e strategie.
CRISPR Paste
Nel 2022 un altro studio è quello pubblicato su Nature Biotechnology da due ex allievi di Zhang. Essi descrivono un metodo consistente nell’affiancare o fondere un terzo elemento, una ricombinasi a serina, LSR, al prime editing per ottenere un sistema a tre blocchi (paste): recombinasi, retrotrascrittasi e l’enzima programmabile modificato per scalfire un solo filamento anziché rescinderli entrambi (nickasi). In tal modo l’editing non risulta più solo una correzione di refusi ma diventa scrittura vera e propria del DNA attraverso tre passaggi:
a) CRISPR va a bersaglio, posizionandosi come un cursore nel punto giusto;
b) la trascrittasi inversa inserisce una breve sequenza ;
c) la ricombinasi riconosce il sito e inserisce la sua integrazione.
La tripletta enzimatica, è in definitiva, costituita da una variante di CRISPR accessoriata con due effettori, dove il primo prepara il terreno al secondo.
Inoltre, il grande vantaggio delle ricombinasi è che catalizzano l’inserzione di lunghi pezzi di DNA senza effettuare il taglio della doppia elica e senza fare affidamento sui naturali sistemi di riparazione delle cellule per saldare la lesione.
Evitare il taglia – incolla è auspicabile, perché causerebbe mutazioni indesiderate e riarrangiamenti cromosomici. Tuttavia, c’è anche da tenere in considerazione alcuni svantaggi che risiedono nelle ricombinasi naturali a causa della loro scarsa efficienza e programmabilità, caratteristica che ha, invece, reso CRISPR molto popolare, dovuta all’RNA guida sintetizzata su misura per condurre il sistema di editing nel punto desiderato.
Nel loro insieme i tre elementi rappresentano uno strumento nuovo denominato PASTE che sta per l’acronimo “Programmable Addition via site – Specific Targeting Elements” e il taglia – incolla che diventa ora un incolla senza taglio.
Questo strumento a tre blocchi potrebbe rivelarsi utile per la cura di malattie genetiche rare nelle quali non basta correggere un piccolo errore ma per le quali si rende necessario la sostituzione del un gene intero. Tra le ipotesi applicative che sono esplorate figurano la fibrosi cistica, l’emofilia e la mallattia di Huntington dove è necessario rimuovere sequenze ripetute.
Nonostante questi buoni propositi, tale tecnologia che rappresenta l’ultima evoluzione dell’editing genomico per incollare geni interi, al momento non è ancora pronta per essere utilizzata nella clinica ma solo in laboratorio in colture cellulari (linfocitarie ed epatocitarie umane) e all’interno di cavie animali (fegato di topo).
Il twin prime editing o Twin PE
Il Twin PE è un’altra innovativa tecnica di editing pubblicata su Nature Biotechnology nel 2022 dal team del Broad Institute del MIT e dell’Università di Harvard sempre guidato da Liu, già inventore di diversi strumenti CRISPR di 2° generazione
Si tratta di un metodo indipendente dalle rotture a doppio filamento, che utilizza una proteina prime editor e due RNA guida di editing primario (pegRNA) per introdurre sequenze più grandi di DNA in posizioni specifiche del genoma con pochi sottoprodotti indesiderati, che può anche diventare con un ulteriore sviluppo, una tecnologia potenzialmente utilizzabile come nuova forma di terapia genica per inserire geni terapeutici in modo sicuro e altamente mirato per sostituire i geni mutati o mancanti.
Questi team di ricercatori dimostrano il potenziale terapeutico di twinPE modificando nelle cellule umane un gene legato alla sindrome di Hunter, una rara malattia genetica causata da un’inversione di uno specifico tratto di DNA lungo 40.000 coppie di basi. TwinPE è stato utilizzato per introdurre un’inversione di una lunghezza simile nello stesso sito nel genoma, mostrando come questo strumento di editing può essere utilizzato per correggere la mutazione che causa la malattia.
L’approccio affronta una limitazione del sistema di prime editing originale, che può modificare solo diverse dozzine di coppie di basi. Tuttavia, lo studio o il trattamento di alcune malattie genetiche richiede modifiche più grandi e come il metodo di prime editing, anche twinPE non recide completamente la doppia elica del DNA contemporaneamente nella stessa posizione, perché ciò può condurre a risultati di editing scarsamente controllati e a dannose anomalie cromosomiche
Alcuni recenti miglioramenti apportati alla tecnologia di prime editing hanno aumentato la sua efficienza, tanto da avvicinarla ad applicazioni terapeutiche, anche se l’editing di sequenze più lunghe di 100 coppie di basi rimane ancora inefficiente. Pertanto, il montaggio di Twin Pe colma questa lacuna.
Come già espresso, il sistema utilizza una proteina editor prima e due RNA guida di editing primari, che guidano il macchinario di editing e codificano le modifiche. Ciascuno dei due RNA guida dirige la proteina di editing per creare un nick a singolo filamento nel DNA in diversi siti mirati nel genoma, evitando il tipo di rottura del doppio filamento che crea sottoprodotti indesiderati in altri metodi. Si sintetizzano due nuovi filamenti di DNA complementari che contengono la sequenza desiderata tra i due nick. Adoperando questo approccio, si inseriscono, si sostituiscono o si o eliminano sequenze lunghe fino a circa 800 coppie di basi.
Per modificare sequenze ancora più grandi, si utilizza un sistema di editing gemello allo scopo di installare “siti di atterraggio” nel genoma per enzimi chiamati ricombinasi sito-specifiche, che catalizzano l’integrazione del DNA in siti specifici nel genoma. Le cellule si trattano con un enzima ricombinasi e poi si introducono lunghi pezzi di DNA nel genoma. La combinazione di enzimi twinPE e ricombinasi permette di modificare sequenze lunghe migliaia di coppie di basi, la lunghezza di interi geni.
Questo studio, può l’inizio di una nuova generazione di strategie di terapia genica.
Caspasi
Le ultime sorprendenti forbici CRISPR, sono ancora rappresentate da un’altra nuova tecnologia in grado di indirizzare l’editing genetico CRISPR a modificare le proteine: una caspasi guidata da CRISPR. Queste caspasi non toccano il DNA e costituiscono delle alternative più sicure del sistema CRISPR standard con un potenziale per eventuali applicazioni del prossimo futuro.
Si tratta di un complesso effettrice di tipo III noto come gRAMP/Cas7-11 che prende di mira l’RNA e si associa ad una proteina simile alla caspasi, TPR- CHAT/ Csx29, per formare uno strumento denominato Craspase, che è guidato da CRISPR.
Craspase taglia specifici filamenti di RNA che attivano gli enzimi proteasi che abbattono le proteine. Al momento, Craspase prende di mira solo specifiche proteine batteriche ma in futuro può essere modificata per altri scopi.
CRISPR-Cas14

Un’altra variante del sistema CRISPR-Cas scoperta nel 2018 dal gruppo coordinato da J. Doudna. è rappresentata da Cas -14 che beneficia di essere più piccola e più semplice rispetto alle altre versioni (Cas 9 e Cas 12) (Fig. 23). Questo nuovo sistema, è probabile che possa essere utilizzato in diversi campi di ricerca come la diagnosi i infezioni virali e microbiche, nonché per il rilevamento e la modifica delle cellule tumorali.
Le applicazioni di CRISPR – Cas su organismi vegetali e animali
Con la tecnica alla base del CRISPR – Cas9, oggi è possibile intervenire all’interno del genoma di organismi vegetali, animali e umani.
In ambito vegetale, le modificazioni di parti della sequenza del DNA rendono molto più preciso e rapido lo sviluppo di varietà di piante agricole più resistenti agli agenti patogeni e alle diverse condizioni ambientali e climatiche, più produttive, più ricche di principi nutritivi e, dunque, in grado di nutrire una popolazione sempre più in crescita.
Servendosi di questo editing genomico, ad esempio, è stato possibile creare un particolare tipo di seme di cotone, privo di aldeide, sostanza tossica per l’uomo, che ha sempre impedito la commestibilità di questa pianta e ricco di proteine e di fibre. E continuano le sperimentazioni per rendere anche la vite più resistente alle infezioni fungine.
Sempre nell’ambito della ricerca sugli organismi vegetali, la modifica, per mezzo del sistema CRISPR, del genoma di una particolare microalga fino a raddoppiarne il contenuto lipidico, la rende una soluzione conveniente e sostenibile come possibile fonte di biocarburanti.
Un ulteriore vantaggio del sistema CRISPR sta nel fatto che se con le tecniche di ingegneria genetica del passato era difficile ottenere più di una singola alterazione del genoma e, quindi, rendere una specie vegetale resistente a più di un agente patogeno o fattore ambientale, oggi si apre tutto un nuovo ventaglio di possibilità.
Relativamente agli organismi animali, al momento le principali applicazioni della proteina Cas9 sembrano essere quelle per l’introduzione di resistenze a malattie di origine virale attraverso la modificazione mirata di geni dell’animale.
gene drive
Un’altra applicazione di CRISPR è uno strumento chiamato gene drive. Ideato originariamente dal biologo statunitense dell’Università di Harvard, Kevin Esvelt, un gene drive è un segmento sintetico di DNA che include il gene Cas9 e un gene guideRNA, insieme a un particolare gene di interesse (chiamato anche payload DNA), il tutto in un’unità auto-funzionante. Il payload DNA può essere un nuovo gene o una versione modificata di un gene esistente.
Una volta che un gene drive viene introdotto nel cromosoma di un organismo diploide, il drive genererà un complesso Cas9/RNAguida che taglierà il cromosoma omologo e quindi copierà il gene drive nella rottura tramite HDR. Con il gene drive ora presente in entrambi i cromosomi, l’organismo diventa omozigote per il drive, compreso il payload DNA.
I gene drive sono in grado di inserire (knocking-in) o disattivare (knocking-out) i geni di un organismo. Ancora più importante, i gene drive possono essere propagati in un’intera popolazione di organismi tramite la riproduzione sessuale, consentendo così la modifica genetica a livello di popolazione.
Un esempio di gene drive proviene da uno studio del 2018 per il controllo della malaria. In questo studio i ricercatori sono riusciti ad ingegnerizzare un gene drive che colpisce esclusivamente le zanzare femmine di Anopheles gambiae, responsabili delle punture e della trasmissione della malaria. Modificando il gene doublesex, una specie di interruttore genetico per la determinazione del sesso dell’insetto, hanno ottenuto una zanzara femmina che presenta caratteristiche morfologiche intermedie tra i due sessi, quindi sterile e incapace di pungere. In questo modo, è possibile fare nascere solo gli esemplari del sesso che non punge, facendo così collassare la riproduzione di popolazioni di insetti vettori della malattia.
Il gene drive potrebbe anche essere utilizzato per eradicare altre malattie trasmesse dagli insetti, come il virus Zika, o eliminare parassiti infestanti per creare colture più efficienti.
Le applicazioni in ambito diagnostico e terapeutico
Dal 2012 anno della sua scoperta, la comunità scientifica mondiale ha iniziato a studiare le potenziali applicazioni del CRISPR – Cas9 in ambito clinico (diagnostico e terapeutico), a cominciare dalle malattie genetiche quali distrofia muscolare e fibrosi cistica, dalle malattie neurologiche come Alzheimer e Parkinson, fino ad arrivare, anno dopo anno, alle malattie infettive come l’AIDS e a particolari tipologie di tumori.
A differenza delle applicazioni sugli organismi animali e vegetali, le pratiche dell’editing genomico sull’organismo umano sono ancora agli albori e per la maggior parte si svolgono sul piano della ricerca e dello studio in laboratorio.
Quello che è chiaro è che, oggi, grazie alle tecniche del più recente sistema di editing genomico, si possono ricreare in vitro sistemi cellulari in grado di “mimare” alcune patologie, con l’obiettivo di identificare in modo più accurato e meticoloso nuove terapie. E in modo più rapido e semplice rispetto al passato.
Si può, inoltre, affermare che, allo stato attuale, CRISPR- Cas9, permettendo di modificare il genoma di una cellula intervenendo direttamente sulla sequenza di DNA patologica, rappresenta l’approccio più avanzato di terapia genica dove le nuove sequenze di DNA che vengono inserite sono date da copie corrette del gene prodotte in laboratorio.
In particolare, nella ricerca sui tumori, CRISPR – Cas9 rappresenta uno strumento rivoluzionario, in quanto ha permesso di velocizzare l’intero processo attraverso il quale, in laboratorio, vengono studiate, su cellule animali, le mutazioni che portano alla malattia. Ma la scelta di usare questo strumento nella ricerca sul tumori è legata anche a questioni di precisione e di puntualità, che consentono di ricreare, in laboratorio e con buona approssimazione, le dinamiche proprie della patologia.
I nuovi studi clinici di editing genetico
Il 2022 è stato un anno molto impegnativo nel campo dell’editing genetico, con numerosi studi clinici avviati per candidati terapeutici di modifica genetica e la progressione di una serie di studi in corso con dei segnali incoraggianti di sicurezza ed efficacia. Alcuni progressi chiave riguardano gli studi di modifica genetica per alcuni tumori, emoglobinopatie e malattie rare.
L’EMA (European Medicines Agency, ossia l’ente regolatorio del Farmaco presso l’Unione Europea), in questo periodo, sta valutando la domanda per l’immissione in commercio del primo farmaco, prodotto dalla Società statunitense Vertex Pharmaceuticals, basato su CRISPR che prende il nome di exagamglogene autotemcel (exa-cel) e che dovrà servire a curare l’anemia falciforme e la beta talassemia trasfusione dipendente. Questa terapia si basa su CRISPR /Cas9 che modifica le cellule staminali di un paziente per riattivare la produzione di emoglobina fetale o HbF. Questa emoglobina è altamente espressa e critica durante lo sviluppo fetale e quindi rapidamente soppressa nelle prime fasi della vita. La sua riattivazione è emersa come strategia molto interessante per trattare l’anemia falciforme e la beta – talassemia compensando il deficit di emoglobina funzionale nell’adulto, osservata in queste due patologie facenti parte di quella famiglia di malattie del sangue, le emoglobinopatie. Se approvata, exa-cel diventerà la prima terapia CRISPR a raggiungere l’approvazione normativa.
Anche per l’angioedema ereditario (HAE), malattia genetica rara caratterizzata da attacchi infiammatori gravi, ricorrenti e imprevedibili con gonfiore negli organi e nei tessuti in tutto il corpo si sta progettando, da parte della Società di Biotecnologie statunitense Intellia Therapeutics, una terapia in vivo basata su CRISPR/Cas 9 denominata NTLA – 2002 che è in fase di valutazione in Nuova Zelanda. La callicreina plasmatica è una proteina conosciuta per guidare molti percorsi infiammatori, compresa la produzione del mediatore infiammatorio bradichinina, sovraprodotto nell’HAE. L’inibizione della callicreina plasmatica è tra gli attuali approcci terapeutici all’HAE. NTLA-2002 è stato specificamente progettato per eliminare il gene codificante la callicreina B1 nelle cellule epatiche per ridurre in maniera permanente l’attività plasmatica della callicreina allo scopo, quindi, di arrestare la produzione di bradichinina.
La stessa Società, in collaborazione con Regeron Pharmaceuticals, sta valutando in uno studio clinico su candidati un’altra strategia terapeutica CRISPR/Cas9 NTLA-2001, questa volta per l’amiloidosi da transitiretina (ATTR), malattia rara e progressiva in cui una proteina nota come TTR diventa mal ripiegata e si accumula come placche nei tessuti di tutto il corpo.
Un’altra Società Farmaceutica statunitense, la Verve Therapeutics, ha rivelato lo scorso anno di aver trattato il primo paziente per l’ipercolesterolemia familiare eterozigote (He FH), con un nuovo farmaco sperimentale, Verve -101, progettato per essere un trattamento a ciclo singolo che disattiva permanentemente il gene PCSK9 nel fegato per ridurre il colesterolo lipoproteico a bassa densità (LDL-C) che guida la malattia.
Interessante sarà sapere cosa succederà nel campo dell’editing genetico nel 2023 dove le sperimentazioni cliniche saranno organizzate nelle aree degli antibatterici, delle malattie del sangue e oculari, dei tumori, dei disordini metabolici e di altre condizioni genetiche. Al di là delle potenzialità straordinarie di questo metodo e degli scenari inediti che apre soprattutto in ambito diagnostico e terapeutico, pesano alcune ombre che, ad oggi, frenano le sperimentazioni cliniche sull’uomo, sebbene queste hanno iniziato comunque a farsi sempre più spazio e a divenire più urgenti. La ricerca, in ogni caso, sta lavorando al superamento di alcuni limiti lasciando ben sperare nella loro soluzione negli anni a venire.
Conclusioni
Oggi, Feng Zhang viene soprannominato il re Mida dei metodi per la sua capacità di ideare, di far funzionare nuove tecnologie dal loop genetico a CRISPR.
Un altro padre di CRISPR è il suo ex mentore Geoge Church, il cui contributo è rappresentato dagli importanti avanzamenti, in questo decennio, nel sequenziamento, nella manipolazione e nella descrizione del DNA. Di Church si dice che abbia il talento di trasformare la fantascienza in scienza. Una delle imprese in cui è impegnato è di editare il genoma di maiale per ottenere organi trapiantabili nell’uomo.
Il terzo nome di questa rosa è di David Liu originario di Taiwan. Nel suo laboratorio le forbici genetiche di CRISPR si sono trasformate in correttori automatici come quelli che utilizziamo quando scriviamo al computer.
Bibliografia:
- Bradley A. (1991). Modifying the mammalian genome by gene targeting. Curr Opin Biotechnol. 1991 Dec;2(6):823 – 9. Review.
- Moore, J.K. and Haber, J.E. (1996). Cell cycle and genetic requirements of two pathways of non-homologous end joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol. Cell. Biol., 16 (5):2164 – 2173.
- Chevalier BS, Kortemme T, Chadsey MS, Baker D, Monnat RJ, Stoddard BL ( 2002). “Design, activity, and structure of a highly specific artificial endonuclease“. Molecular Cell. 10 (4): 895–905
- Van der Oost J., (2013).” Molecular biology. New tool for genome surgery” Science 339: 768-770.
- Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD (2010). “Genome editing with engineered zinc finger nucleases”. Nat Rev Genet. 2010 Sep ;11(9):636-46.
- Hsu PD, Lander ES, Zhang F. (2014). “Development and applications of CRISPR-Cas9 for genome engineering“. Cell. 157 (6): 1262–1278.
- Wright AV, Nuñez JK, Doudna JA ( 2016) . “Biology and Applications of CRISPR Systems: Harnessing Nature’s Toolbox for Genome Engineering“. Cell. 164 (1–2): 29–44
- Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A (1987). “Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product“. J Bacteriol. 169 (12): 5429–33
- Mojica FJ, Díez-Villaseñor C, Soria E, Juez G.( 2000) .”Biological significance of a family of regularly spaced repeats in the genomes of archaea, bacteria and mitochondria”. Mol Microbiol 36:244–246.
- Jansen R, Embden JD, Gaastra W, Schouls LW.( 2002) .”Identification of genes that are associated with DNA repeats in prokaryotes”. Mol Microbiol 43:1565–1575
- Mojica FJM, Díez-Villaseñor C, García-Martínez J, Soria E. (2005). “Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements.” J Mol Evol 60:174–182.
- Barrangou R, et al.(2007) “CRISPR provides acquired resistance against viruses in prokaryotes.” Science 315 : 1709-1712.
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier EA.( 2012). “A programmable dual-RNA-guided DNA endonuclease inadaptive bacterial immunity”. Science 337:816–821.
- Jinek M., East A., Cheng A., Lin S., Ma. E. and Doudna J. (2013) “RNA-programmed genome editing in human cells“ Elife. Jan 29;2:e00471
- Mali P., Yang L., Esvelt K.M., Aach J. et al. ( 2013). “RNA-guided human genome engineering via Cas 9”. Science 339: 823-826
- Cong L., Ran F.A., Cox D. (2013) “Multiplex Genome Engineering using CRISPR/Cas Systems“ Science 339 : 819 – 823
- Gaudelli N.M., Komor A.C., Rees H.A. et al. (2017) “Programmable base editing of A-T to G-C in genomic DNA without DNA cleavage“ Nature 551: 464- 471
- Cox D.B.T., Gootenberg J.S., Abudayyeh O.O. et al (2017) “RNA editing with CRISPR -Cas 13“ Science, 358: 1019-1027
- Anzalone A.V., Randolph P.B., Davis J.R. et al ( 2019) “Search and replace genome editing without double strand breaks or donor DNA“ Nature 576: 149-157
- Mazzaracca R. “CRISPR: dalle origini al ‘prime editing” , Osservatorio Terapie Avanzate , 28 Ottobre 2019
- Meldolesi A. (2023) “Dai vaccini all’editing genomico, il futuro è nelle gocce di grasso” Osservatorio Terapie avanzate, 22 febbraio 2023
- Meldolesi A. (2022) “Crispr-paste: la combinazione a tre per incollare geni interi” Osservatorie Terapie avanzate, 14 Dicembre 2022.
- Liu X., Zhang L., Wang H. et al.(2022) “Target RNA activates the protease activity of CRASPASE to confer antiviral defense“ Molecular Cell 82: 4503 – 4518
- Hu C., van Beljouw S.P.B., Nam K.H et al (2022) “Craspase is a CRISPR RNA – guided, RNA activated protease“ Science 377: 1278 – 1285.
- Anzalone A.V., Gao X.D., Podracky C.J. et al. (2022) “Programmable deletion, replacement, integration and inversion of large DNA sequences with twin prime editing” Nature Biotechnology 40: 731-740
- Harrington L.C., Burstein D., Cheng J.S. et al (2018) “Programmed DNA destruction by miniature CRISPR-Cas14 enzymes” Science 362:839 – 842.