Domani l’ente regolatorio farmaceutico statunitense, la FDA, deciderà se approvare l’uso del vaccino Pfizer-Biontech negli USA. Questo post è un breve riassunto della RICHIESTA fatta dalla casa farmaceutica americana Pfizer in collaborazione con l’azienda tedesca Biontech.
Pfizer e BioNTechhanno presentato una domanda di autorizzazione all’uso di emergenza per un vaccino sperimentale destinato a prevenire la malattia da Coronavirus 2019 (COVID-19) causata da SARS-CoV-2; questa richiesta è stata presentata il 20 novembre 2020. Il vaccino è basato su un antigene SARS-CoV-2, la cosiddetta glicoproteina spike, codificato da RNA formulato in nanoparticelle lipidiche.
LA RICHIESTA DI APPROVAZIONE DEL VACCINO
In risposta all’attuale crisi sanitaria globale, lo sviluppo del vaccino Pfizer-BioNTech COVID-19 ha garantito i più alti standard di conformità e qualità, procedendo rapidamente per affrontare questa esigenza medica urgente e insoddisfatta.
Lo “studio C459100” è uno studio registrativo di fase 1/2/3 in corso, randomizzato e controllato con placebo. È stato avviato come studio di fase 1/2 negli Stati Uniti, quindi è stato modificato per estenderlo a uno studio globale di fase 2/3 che ha arruolato circa 44.000 partecipanti per una valutazione tempestiva e ben potenziata degli endpoint di sicurezza, immunogenicità ed efficacia ed ha coinvolto anche adolescenti dai 12 ai 17 anni di età. Questo studio viene condotto in luoghi selezionati in tutto il mondo per garantire la diversità della popolazione iscritta.
VACCINO VS PLACEBO
I dati di circa 38.000 partecipanti randomizzati 1:1 a ricevere vaccino o placebo con una mediana di 2 mesi di follow-up dopo la Dose 2 del regime vaccinale a 2 dosi nello “Studio C4591001” mostrano che il vaccino “BNT162b2” ad un dosaggio di 30 μg era sicuro e ben tollerato nei partecipanti di età ≥16 anni.
L’incidenza di eventi avversi gravi e decessi è stata bassa nel contesto del numero di partecipanti arruolati e comparabile per BNT162b2 e placebo. Anche l’incidenza delle interruzioni dovute ad eventi avversi è stata generalmente bassa e simile tra i gruppi BNT162b2 e placebo.
Il regime a due dosi del ‘vaccino BNT162b2’ ha suscitato solidi titoli di neutralizzazione SARS-CoV-2 e livelli di IgG leganti S1 (cioè la proteina spike) sia negli adulti più giovani che in quelli più anziani. Dopo la seconda dose di vaccino e fino a circa il Giorno 50 (1 mese dopo la dose 2), i titoli medi di neutralizzazione di SARS-CoV-2 erano paragonabili o superiori al titolo della media geometrica di un siero di convalescenza umano recuperati da individui che avevano passato la malattia COVID-19. È in corso un’ulteriore valutazione della persistenza degli anticorpi. I dati dello studio BNT162-01 dimostrano che il vaccino BNT162b2 ha anche suscitato forti risposte CD4+1 e forti risposte delle cellule T CD8+.
Un totale di 10 casi di COVID-19 grave si sono verificati dopo la dose 1, 1 nel gruppo BNT162b2, rispetto ai 9 casi nel gruppo placebo.
Tra tutti i partecipanti (indipendentemente dall’evidenza di infezione prima o durante il regime di vaccinazione), 50 casi di COVID-19 si sono verificati dopo la Dose 1 nel gruppo BNT162b2 rispetto ai 275 casi nel gruppo placebo, indicando un’efficacia vaccinale stimata dell’82%.
UNA PRECOCE PROTEZIONE
Una scheda sul sondaggio riguardo ad un possibile vaccino anti-covid.
L’inizio precoce della protezione è immediatamente evidente dalle curve di incidenza cumulativa, che mostrano che l’insorgenza della malattia segue congiuntamente BNT162b2 e placebo fino a circa 14 giorni dopo la Dose 1, a quel punto le curve divergono, con casi in costante accumulo nel gruppo placebo, pur rimanendo praticamente piatto nel gruppo BNT162b2.
Il vaccino Pfizer-BioNTech COVID-19, BNT162b2 (30 μg), viene somministrato per via intramuscolare (IM) come una serie di due dosi da 30 μg della soluzione di vaccino diluita (0,3 mL ciascuna) secondo il seguente schema: una singola dose seguita da una seconda dose.
Il vaccino BNT162b2 è fornito come flaconcino a dose multipla (5 dosi) contenente una sospensione congelata (tra -80ºC e -60ºC priva di conservanti. BNT162b2 deve essere scongelato e diluito nella sua fiala originale con 1,8 mL di soluzione iniettabile sterile di cloruro di sodio allo 0,9% prima della somministrazione. Dopo la diluizione, i flaconcini multidose devono essere conservati tra 2°C e 25°C e utilizzato entro 6 ore dal momento della diluizione.
I dati disponibili da altri coronavirus come SARS e MERS hanno stabilito che gli anticorpi contro la proteina S (spike) possono bloccare il legame del virus alle cellule e prevenire l’infezione virale.
Dopo l’iniezione, gli le nanoparticelle lipidiche vengono assorbite dalle cellule e l’RNA viene rilasciato nel citosol. Nel citosol, l’RNA viene tradotto nella proteina virale codificata (la proteina spike). L’antigene P2 della proteina spike si incorpora nelle membrane cellulari e induce una risposta immunitaria adattativa. Poiché la proteina spike è l’antigene che riconosce il recettore dell’ACE2 e consente l’infezione delle cellule ospiti, è un bersaglio chiave degli anticorpi neutralizzanti il virus. Inoltre, poiché la proteina spike espressa dall’RNA è frammentata a livello intracellulare, i peptidi risultanti possono essere presentati sulla superficie cellulare, innescando una risposta immunitaria mediata dalle cellule T specifica con attività contro il virus.
TOLLERANZA AL VACCINO
La somministrazione del vaccino BNT162b2 mediante iniezione intramuscolare in topi maschi e femmine una volta alla settimana, per un totale di 3 cicli settimanali di dosaggio (30 μg e 100 μg), è stata tollerata senza evidenza di tossicità sistemica negli studi di tossicità in presenza di una robusta risposta immunitaria.
La fase 3 (che è in corso) includeva analisi ad interim pianificate del primo endpoint primario di efficacia, valutazioni di efficacia e sicurezza in corso, inclusa la valutazione della reattogenicità in un sottogruppo di partecipanti e la valutazione esplorativa dell’immunogenicità del vaccino in un sottogruppo di partecipanti. La fase 3 è in corso presso siti negli Stati Uniti, Brasile, Argentina, Turchia, Sud Africa e Germania. I partecipanti sono stati stratificati per fascia di età.
ANALISI DI EFFICACIA
L’analisi di efficacia finale doveva essere condotta quando almeno il numero totale prespecificato di 164 eventi di efficacia si era accumulato. La sicurezza e la persistenza a lungo termine del follow-up dell’efficacia continueranno per almeno 2 anni e / o alla fine dello studio. Le analisi di sicurezza ed efficacia hanno incluso i 360 partecipanti che sono stati analizzati per la Fase 2.
Quando ci sarà la piena approvazione normativa di un vaccino nei paese partecipanti allo studio, il vaccino BNT162b2 sarà offerto a tutti i partecipanti che avevano assunto il placebo. Riconosciamo che potremmo essere obbligati a farlo prima, in conformità con il comitato etico, le linee guida cliniche e normative, poiché le analisi intermedie e finali pianificate dell’efficacia sono state completate con travolgente successo.
In tutti i casi, intendiamo seguire i partecipanti allo studio di fase 3 in corso fino all’originale pianificato 24 mesi dopo la vaccinazione, indipendentemente dal fatto che i partecipanti optino per il passaggio dal placebo alla vaccinazione attiva.
REAZIONI LOCALI E GENERALIZZATE ALLA VACCINAZIONE
Le reazioni locali sono state generalmente poco frequenti nei destinatari del placebo. La maggior parte delle reazioni locali nei gruppi BNT162b2 sono state di gravità lieve o moderata e si sono risolte entro diversi giorni dall’esordio. Non sono state segnalate reazioni potenzialmente pericolose per la vita. Il dolore al sito di iniezione è stata la reazione locale indotta più frequentemente per numero di dosi, mentre l’arrossamento ed il gonfiore sono stati infrequenti.
Gli eventi sistemici erano generalmente poco frequenti nei destinatari del placebo. Gli eventi sistemici suggeriti generalmente aumentavano in frequenza e / o gravità con l’aumento del livello di dose e del numero di dosi di BNT162b2. Anche l’uso di farmaci antipiretici / antidolorifici è aumentato in frequenza con l’aumento del livello di dose e del numero di dosi. La maggior parte degli eventi sistemici sono stati lievi o moderati, si sono verificati entro i primi 1-4 giorni dalla somministrazione e si sono risolti entro 1-3 giorni dall’esordio. Non sono stati segnalati eventi di grado 4 (potenzialmente pericolosi per la vita). Gli eventi sistemici indotti più frequenti per numero di dosi e livelli di dose in entrambi i gruppi di età sono stati affaticamento (dall’8% al 75%), mal di testa (dallo 0% al 67%), brividi (dallo 0% al 58%) e dolore muscolare (0 dal% al 58%). La febbre era rara (dallo 0% al 17%).
Tra tutti i 43.448 partecipanti arruolati, dalla dose 1 alla data di cutoff dei dati, le proporzioni dei partecipanti che hanno riportato almeno 1 Serio Evento Avverso erano simili nel gruppo del vaccino BNT162b2 (0,6%) e nel gruppo placebo (0,5%).
Il protocollo aveva regole di interruzione prespecificate che includevano il monitoraggio di casi gravi di COVID-19 e questi criteri di interruzione non erano soddisfatti. Il confinamento della maggior parte dei casi gravi ai gruppi placebo suggerisce che non vi sono prove di malattia potenziata associata al vaccino (VAED) inclusa la malattia respiratoria potenziata associata al vaccino (VAERD).
PROTEZIONE
L’inizio precoce della protezione è immediatamente evidente nella Figura (la 13), che mostra l’incidenza cumulativa per la prima insorgenza di COVID-19 dopo la Dose 1 tra tutti i partecipanti vaccinati in base alla popolazione di Dose 1 con efficacia totale (intenzione di trattamento modificata). L’esordio della malattia sembra seguire insieme per BNT162b2 e placebo fino a circa 14 giorni dopo la Dose 1, a quel punto le curve divergono, con casi in costante accumulo nel gruppo placebo, e diversamente rimanendo praticamente invariati nel gruppo BNT162b2.
Dopo 7 giorni dalla seconda dose, il Covid-19 si è manifestato in 8 soggetti (su 18.198) del gruppo dei vaccinati e in 162 soggetti (su 18.325) del gruppo controllo sottoposti a placebo, mostrando un’efficacia del vaccino del 95%.

 In risposta all’attuale crisi sanitaria globale, lo sviluppo del vaccino Pfizer-BioNTech COVID-19 ha garantito i più alti standard di conformità e qualità, procedendo rapidamente per affrontare questa esigenza medica urgente e insoddisfatta.
In risposta all’attuale crisi sanitaria globale, lo sviluppo del vaccino Pfizer-BioNTech COVID-19 ha garantito i più alti standard di conformità e qualità, procedendo rapidamente per affrontare questa esigenza medica urgente e insoddisfatta. I dati di circa 38.000 partecipanti randomizzati 1:1 a ricevere vaccino o placebo con una mediana di 2 mesi di follow-up dopo la Dose 2 del regime vaccinale a 2 dosi nello “Studio C4591001” mostrano che il vaccino “BNT162b2” ad un dosaggio di 30 μg era sicuro e ben tollerato nei partecipanti di età ≥16 anni.
I dati di circa 38.000 partecipanti randomizzati 1:1 a ricevere vaccino o placebo con una mediana di 2 mesi di follow-up dopo la Dose 2 del regime vaccinale a 2 dosi nello “Studio C4591001” mostrano che il vaccino “BNT162b2” ad un dosaggio di 30 μg era sicuro e ben tollerato nei partecipanti di età ≥16 anni.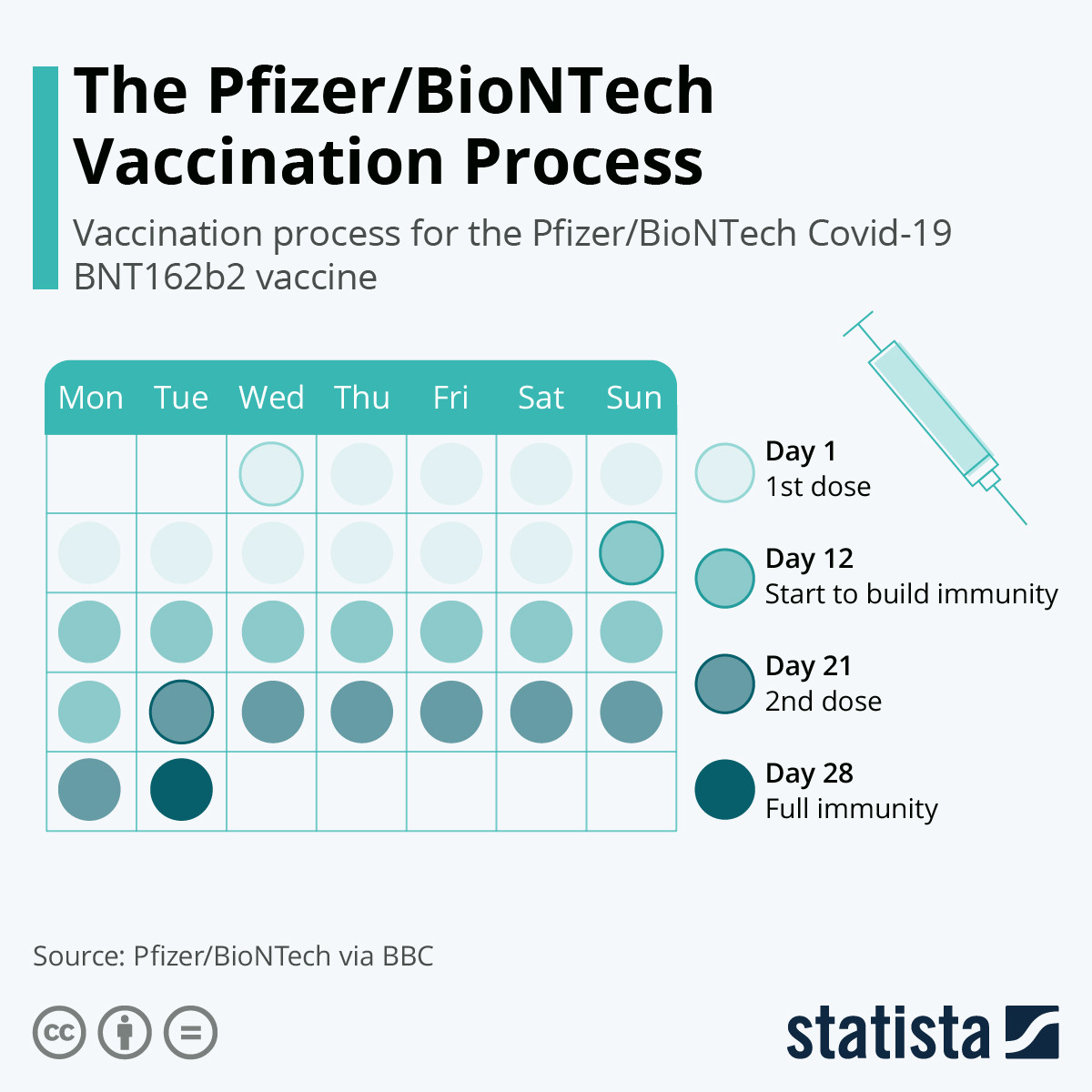


 La somministrazione del vaccino BNT162b2 mediante iniezione intramuscolare in topi maschi e femmine una volta alla settimana, per un totale di 3 cicli settimanali di dosaggio (30 μg e 100 μg), è stata tollerata senza evidenza di tossicità sistemica negli studi di tossicità in presenza di una robusta risposta immunitaria.
La somministrazione del vaccino BNT162b2 mediante iniezione intramuscolare in topi maschi e femmine una volta alla settimana, per un totale di 3 cicli settimanali di dosaggio (30 μg e 100 μg), è stata tollerata senza evidenza di tossicità sistemica negli studi di tossicità in presenza di una robusta risposta immunitaria. L’analisi di efficacia finale doveva essere condotta quando almeno il numero totale prespecificato di 164 eventi di efficacia si era accumulato. La sicurezza e la persistenza a lungo termine del follow-up dell’efficacia continueranno per almeno 2 anni e / o alla fine dello studio. Le analisi di sicurezza ed efficacia hanno incluso i 360 partecipanti che sono stati analizzati per la Fase 2.
L’analisi di efficacia finale doveva essere condotta quando almeno il numero totale prespecificato di 164 eventi di efficacia si era accumulato. La sicurezza e la persistenza a lungo termine del follow-up dell’efficacia continueranno per almeno 2 anni e / o alla fine dello studio. Le analisi di sicurezza ed efficacia hanno incluso i 360 partecipanti che sono stati analizzati per la Fase 2.