GLI AGONISTI DEL RECETTORE DEL GLP-1: BREVE STORIA
- Articolo del dott. Concetto De Luca
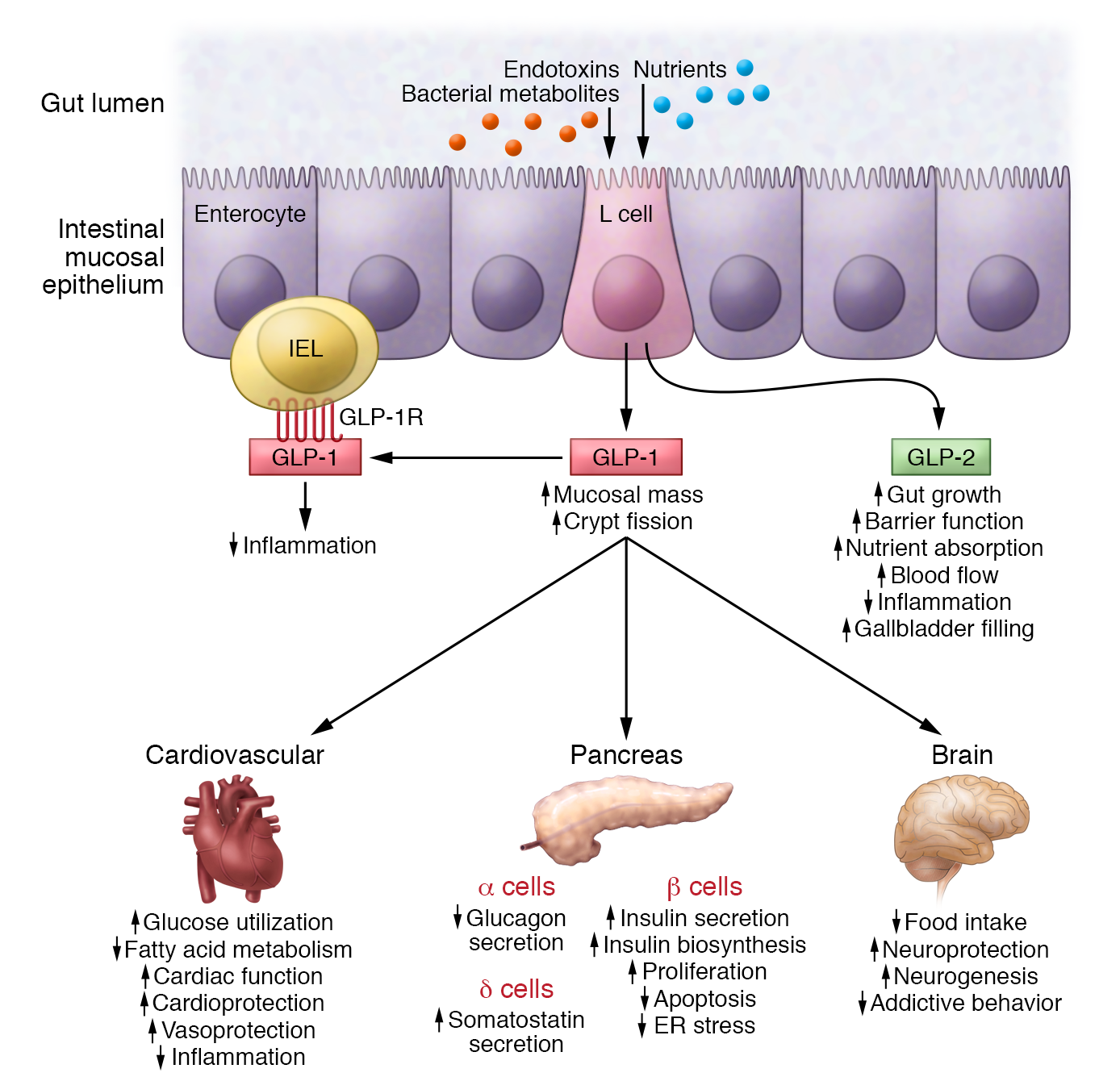
Gli agonisti del recettore del GLP-1 (incretino-mimetici) sono farmaci usati come antidiabetici e studiati per la terapia dell’obesità1.
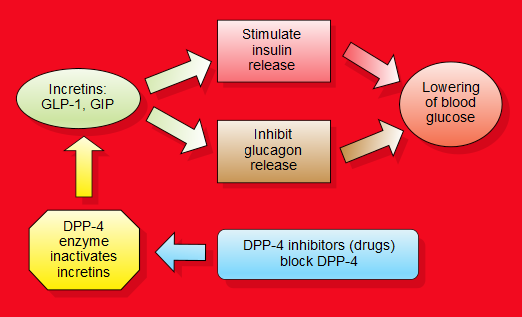
Il GLP-1 (glucagon-like peptide 1) è un ormone prodotto dall’intestino che stimola la secrezione di insulina e inibisce la secrezione di glucagone da parte del pancreas. Il suo rilascio avviene dopo il pasto, entrando quindi in azione solamente quando la glicemia sale per effetto dei carboidrati introdotti col cibo. Il GLP-1 rallenta lo svuotamento gastrico, aumentando il senso di sazietà in risposta all’assunzione di cibo, e riduce l’appetito, agendo direttamente sui centri di regolazione della fame del sistema nervoso centrale. Dopo il suo rilascio il GLP-1 viene rapidamente degradato da uno specifico enzima, la DPP-4 (dipeptil-peptidasi 4), pertanto il suo impiego a scopo terapeutico non è praticabile se non con una infusione continua. Per ovviare al problema della rapida degradazione del GLP-1 sono stati sviluppati degli analoghi, definiti più correttamente “agonisti del recettore del GLP-1”, con struttura più o meno simile al GLP-1, che resistono all’azione di degradazione esercitata dalla DPP-4 e che a volte sono legati a molecole che ne rallentano l’assorbimento sottocutanea2.
SECRETINA ED INCRETINA

L’attività endocrina del tratto gastrointestinale è stata studiata per più di un secolo, con ormoni intestinali come la secretina emersi (19023) dagli studi fondamentali dei fisiologi inglesi William Maddock Bayliss (1860 – 19244) ed Ernest Herny Starling (1866 – 19275)6. La scoperta della secretina fu anche lo sfondo della conferenza crooniana di Starling del 1905, in cui egli coniò la parola ormone (dal greco “hormoa”: suscito all’attività).
Il concetto che l’intestino controllasse anche le secrezioni delle isole pancreatiche è stato supportato da esperimenti che hanno dimostrato che la somministrazione di estratti intestinali grezzi abbassava il glucosio nel sangue negli animali. Lo sviluppo del test radioimmunologico dell’insulina ha consentito la descrizione dell’effetto incretinico7, vale a dire che il glucosio somministrato nell’intestino potenzia la secrezione di insulina in misura maggiore rispetto alla stimolazione isoglicemica della secrezione di insulina ottenuta attraverso la somministrazione endovena di glucosio.
UN FATTORE ORMONALE INTESTINALE

Incretina è una parola e un concetto costruiti per un fattore ormonale intestinale che si ritiene integri la secretina nell’effetto sulla secrezione pancreatica. Pertanto, mentre la secretina stimola la secrezione di acqua e bicarbonato dalle cellule pancreatiche esocrine, venne suggerito fin dall’inizio che uno od altri ormoni intestinali avrebbero stimolato la secrezione interna o endocrina dalle isole pancreatiche.
Dopo la scoperta dell’insulina da parte di Banting & Best nel 19228, furono intrapresi nuovi tentativi per esaminare gli estratti della mucosa duodenale e la loro influenza sulle concentrazioni di glucosio nel sangue9.
Risultati promettenti furono ottenuti nel 1930 dai fisiologi dell’Università di Chicago, Jean La Barre ed Eugene U. Still, i quali riferirono di aver ottenuto, dalla lavorazione in vitro di estratti duodenali, due frazioni interessanti: una con secretina grezza, che in sofisticati esperimenti di circolazione incrociata nei cani stimolava la secrezione da pancreas esocrino e un altro che abbassava le concentrazioni di glucosio nel sangue senza effetti sulla secrezione del pancreas esocrino10.
UN ORMONE CHE STIMOLA IL PANCREAS ENDOCRINO
 Inizialmente essi ipotizzarono che l’effetto ipoglicemizzante fosse dovuto alla stimolazione della secrezione di insulina
Inizialmente essi ipotizzarono che l’effetto ipoglicemizzante fosse dovuto alla stimolazione della secrezione di insulina
Successivamente, nel 1932, La Barre usò per la prima volta la parola “incretina“, indicando un ormone intestinale che stimola il pancreas endocrino, compreso il rilascio di insulina. Egli propose anche che l’incretina avrebbe potuto essere utilizzata come trattamento per il diabete mellito.
Tra il 1939 ed il 1940 l’idea di un ormone incretinico con effetto ipoglicemizzante subì un duro colpo per effetto della Scuola di Endocrinologia dell’Università di Chicago, guidata da Andrew Conway Ivy11 (25 febbraio 1893 – 7 febbraio 1978), noto per la scoperta della colecistochinina (CCK) nel 1928. Dopo tre pubblicazioni in rapida successione nel 1939-40 sull’acidificazione del duodeno nei cani a varie concentrazioni di glucosio nel sangue, Ivy et al. conclusero che l’esistenza di un’incretina fosse molto improbabile.
invenzione del dosaggio radioimmunologico

Essi si sbagliavano, ma le loro pubblicazioni paralizzarono ulteriori idee e iniziative sull’incretina per un quarto di secolo12.
L’anno 1960 vide un importante passo avanti per la biomedicina e non ultima l’endocrinologia. Esso ha virtualmente cambiato il mondo e rivitalizzato l’interesse per l’incretina.
Fu l’invenzione del dosaggio radioimmunologico (RIA) da parte di Solomon Aaron Berson (22 aprile 1918 – April 11, 197213) e Rosalyn Sussman Yalow (19luglio 1921 – 30maggio 201114) 15. La tecnica RIA ha consentito per la prima volta in modo abbastanza semplice, ma accurato, la misurazione delle molecole presenti in concentrazioni da pico a femtomolari.
Individuazione di ormoni peptidici

A quelle concentrazioni circola nel plasma un mondo di sostanze biologicamente attive, compresi gli ormoni peptidici. La possibilità di misurazioni dirette ed affidabili degli ormoni pancreatici e gastrointestinali nel plasma riaprì presto la questione delle incretine.
Nel 1964, Neil McIntyre (1930 – 19luglio 202016) et al.17 a Londra ed H. Elrick et al.18, a Denver, USA, hanno dimostrato in modo indipendente che il glucosio orale provoca una risposta insulinica considerevolmente maggiore rispetto al glucosio endovenoso, anche a concentrazioni di glucosio nel sangue simili. Quindi, l’intestino ospita effettivamente fattori ormonali insulinotropici. E in altre parole, esiste il meccanismo delle incretine.
Il primo ormone incretinico, il polipeptide insulinotropico glucosio-dipendente (GIP, Peptide Inibitorio Gastrico19), fu isolato dal fisiologo statunitense John Brown (1938 – 15ottobre 201620) tra la fine degli anni ‘60 e l’inizio degli anni ’70.
SCOPERTA DEL GIP

Inizialmente il GIP fu individuato come un inibitore della secrezione acida gastrica21 ma, poco dopo, venne dimostrato che esso è un potente liberatore di insulina durante l’iperglicemia22. I seguenti studi quantitativi sull’effetto incretinico del GIP, tuttavia, hanno suggerito che il GIP non poteva spiegare l’intero effetto ormonale intestinale sulla secrezione di insulina dopo glucosio orale.
Poi, tra la metà e la fine degli anni ’80, è emerso un ulteriore ormone intestinale con attività incretinica. Lo sfondo della scoperta furono le osservazioni RIA del medico statunitense Roger H. Unger (7marzo 1924 – 22agosto 202023) e collaboratori24 all’inizio degli anni ’60, secondo cui la mucosa intestinale esprimeva una certa immunoreattività simile al glucagone, che era diversa dal noto peptide del glucagone pancreatico; da qui il nome “glucagone intestinale”25.
bioattività dei peptidi del glucagone intestinale
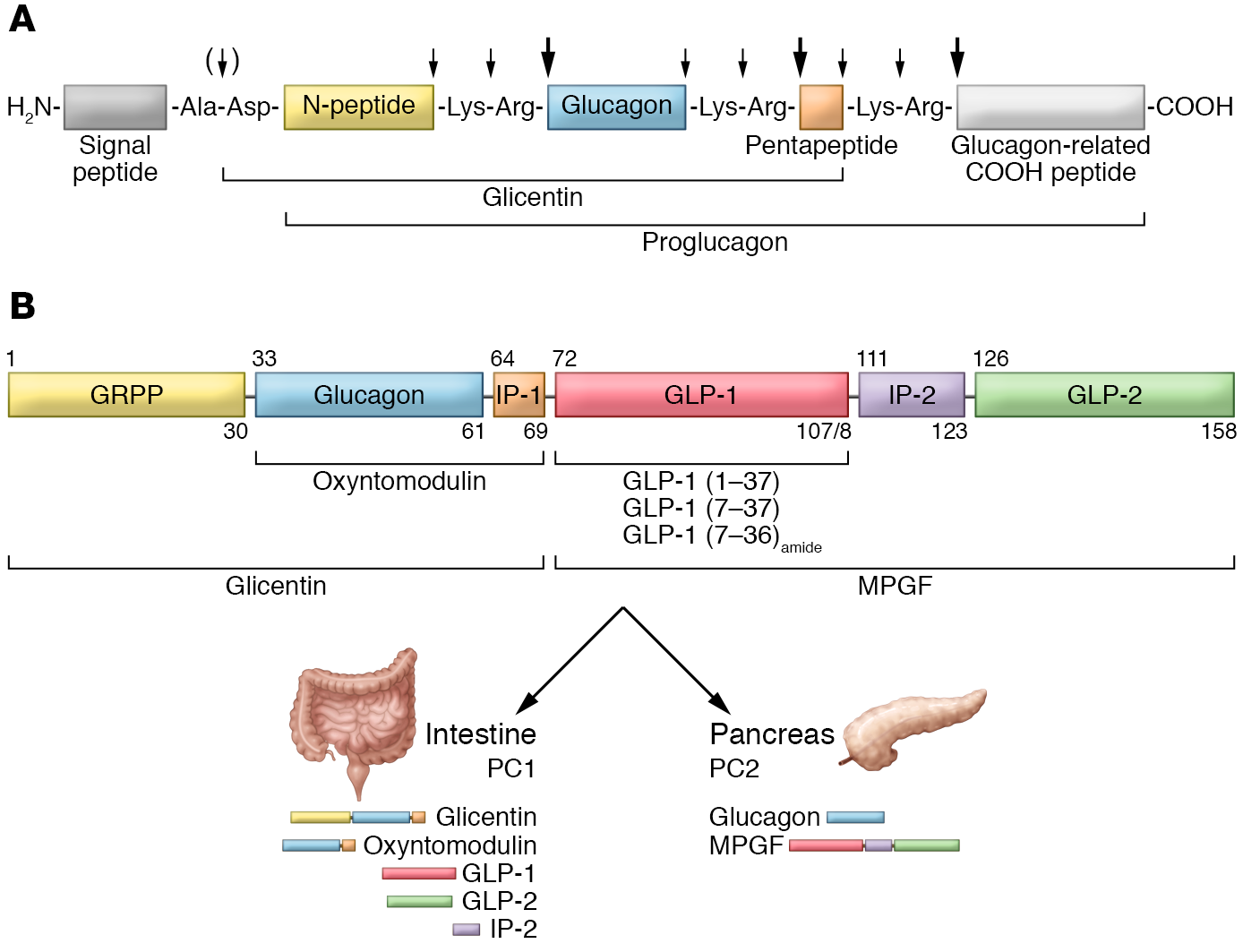
Diversi laboratori in Europa e Nord America hanno successivamente cercato di identificare la bioattività e la struttura dei peptidi del glucagone intestinale. Una premessa essenziale per il successo in questo sforzo divenne la clonazione e il sequenziamento dei geni del preproglucagone dei mammiferi da parte del farmacologo statunitense Graeme Bell e collaboratori nel 198326. Essi determinarono la struttura del preproglucagone pancreatico di criceto dalla sequenza del suo cDNA (DNA complementare).
Mentre il glucagone è un ormone polipeptidico di 29 aminoacidi sintetizzato dalle cellule A del pancreas endocrino, il preproglucagone è composto di 180 aminoacidi e contiene la sequenza del glucagone oltre a due polipeptidi simili al glucagone disposti in tandem. I due nuovi peptidi simili al glucagone furono chiamati GLP-1 e GLP-2. Entrambi i GLP erano espressi nell’intestino.
La purificazione da estratti intestinali nei laboratori di Joel F. Habener e colleghi, ad Harvard, nel 198727, ha mostrato, attraverso la tecnica del DNA ricombinante, che il GLP-1 era sintetizzato anche in una forma troncata con marcate effetto di rilascio dell’insulina28.
GLP-1 troncato

Inoltre, è risultato, soprattutto grazie alle ricerche, oltre che di Habener, della biochimica serba29 trasferitasi negli USA30, Svetlana Mojsov31, dell’endocrinologo canadese Daniel Joshua Drucker32 (nato il 23giugno 195633) e del fisiologo danese Jens Juul Holst34 che il GLP-1 troncato35 [GLP-1 (7-37), GLP-1 (7-37) ed il GLP-1 (7-36)] inibisce anche la secrezione del glucagone pancreatico, che insieme al suo effetto insulinotropico contrasta l’iperglicemia nel diabete36.
All’inizio degli anni novanta, gruppo di Holst si rese conto che un tipo di GLP-1 troncato (proglucagone 78-107-amide) era estremamente interessante e, in studi successivi, dimostrò che esso inibiva fortemente la motilità gastrica e la secrezione esocrina gastrica e pancreatica37, coerente con un ruolo importante di questo ormone come regolatore della funzionalità del tratto gastrointestinale superiore.
regolazione della funzionalità del tratto gastrointestinale superiore
{kind=link}
Hanno inoltre dimostrato che le infusioni di GLP-1 negli esseri umani inibiscono l’appetito e l’assunzione di cibo, azioni successivamente sfruttate in clinica per trattare l’obesità.
Ma sebbene il GLP-1 avesse chiaramente un potenziale terapeutico, le iniezioni sottocutanee di GLP-1 si rivelarono deludenti ed inefficaci. La spiegazione era data dal metabolismo estremamente rapido e dall’inattivazione del GLP-1. Infatti, l’emivita circolante del GLP-1 era solo di 1,5-2 minuti nei soggetti umani con diabete.
Dal veleno di una lucertola il GLP-1R

Nel 1992 l’endocrinologo statunitense John Eng, mentre lavorava presso il Veterans Administration Medical Center nel Bronx, New York, isolò38 dal veleno di lucertola dell’Heloderma awareum un peptide che agisce da agonista del recettore del GLP-1 (GLP-1R). Il suo nome è exenatide (exendin-4 sintetico).
La scoperta dell’azione del Glucagon-like peptide-2 (GLP-239) nel 1995 ha permesso al dott. Daniel J. Drucker, insieme all’Università di Toronto e all’University Health Network, di negoziare un accordo di licenza con Allelix Biopharmaceuticals Inc. che delineava i termini a sostegno della licenza della proprietà intellettuale del GLP-2 e della commercializzazione di Agonisti del recettore GLP-2.
UN FARMACO PER LA SINDROME DELL’INTESTINO CORTO

Dopo diversi anni di validazione preclinica, essi riuscirono ad ampliare il programma di sviluppo e ad intraprendere studi sull’uomo. L’indicazione iniziale selezionata era la SBS (Short Bowel Syndrome), che riflette in parte le azioni note del GLP-2 nell’espandere la superficie della mucosa e migliorare l’assorbimento dei nutrienti, con efficacia dimostrata in molteplici modelli preclinici indipendenti di SBS40.
Un successivo studio pilota di dosaggio di h[Gly2]-GLP-2, teduglutide, è stato condotto su 16 soggetti affetti da SBS, con aumenti osservati nell’assorbimento di energia e ridotta escrezione di energia fecale, senza eventi avversi importanti sulla sicurezza. Teduglutide è stato approvato negli Stati Uniti con il nome Gattex il 21 dicembre 2012, dove gli è stato riconosciuto lo status di farmaco orfano41.
agonisti del GLP-1R
Il programma di sviluppo clinico originale per exendin-4 utilizzava iniezioni due volte al giorno del peptide non modificato. Studi cruciali in soggetti affetti da Diabete tipo 2 hanno infine portato all’approvazione di exenatide (nome commerciale Byetta), il primo agonista del GLP-1R, il 28 aprile 2005, somministrato due volte al giorno. Questi sforzi hanno stimolato lo sviluppo di una forma di exenatide una volta alla settimana in sospensione di microsfere, in definitiva la prima terapia una volta alla settimana approvata per il diabete, nel 2012. Exenatide una volta alla settimana si è rivelato più efficace della preparazione due volte al giorno, con una maggiore riduzione della glicemia e un controllo comparabile del peso corporeo42. Oggi, diversi agonisti del GLP-1R (piccoli peptidi e proteine ad alto peso molecolare) sono approvati per il trattamento del diabete tipo 2 e un singolo farmaco, liraglutide43 (nome commerciale Victoza), è stato approvato per il trattamento dell’obesità nel 2014.
La dipeptidil peptidasi-4

Ispirandosi ai lavori del biochimico tedesco Rolf Mentlein (nato il 18febbraio 1948) a Kiel, Holst e C. F. Deacon, a Copenaghen, hanno dimostrato nel 1995 che la molecola GLP-1 veniva scissa dall’enzima dipeptidil-peptidasi-4 (DPP-4) in vivo e che gli inibitori di questo enzima potevano proteggere completamente la molecola44.
La dipeptidil peptidasi-4 (DPP4 o DPPIV)45, nota anche come proteina complessante l’adenosina deaminasi 2 o CD26 (cluster di differenziazione 26), è una proteina che, nell’uomo, è codificata dal gene DPP446. La DPP-4 è una serina esopeptidasi che scinde i dipeptidi X-prolina o X-alanina dall’N -terminale dei polipeptidi.
L’enzima fu scoperto nel 1966 dai ricercatori finlandesi Väinö K. Hopsu-Havu & George G. Glenner47 e, in seguito a vari studi chimici, fu chiamato dipeptidil peptidasi IV [DP IV]. Sin dalla sua scoperta, la serina proteasi DPP-4 è stata un popolare oggetto di ricerca48.
inibitori del DPP-4
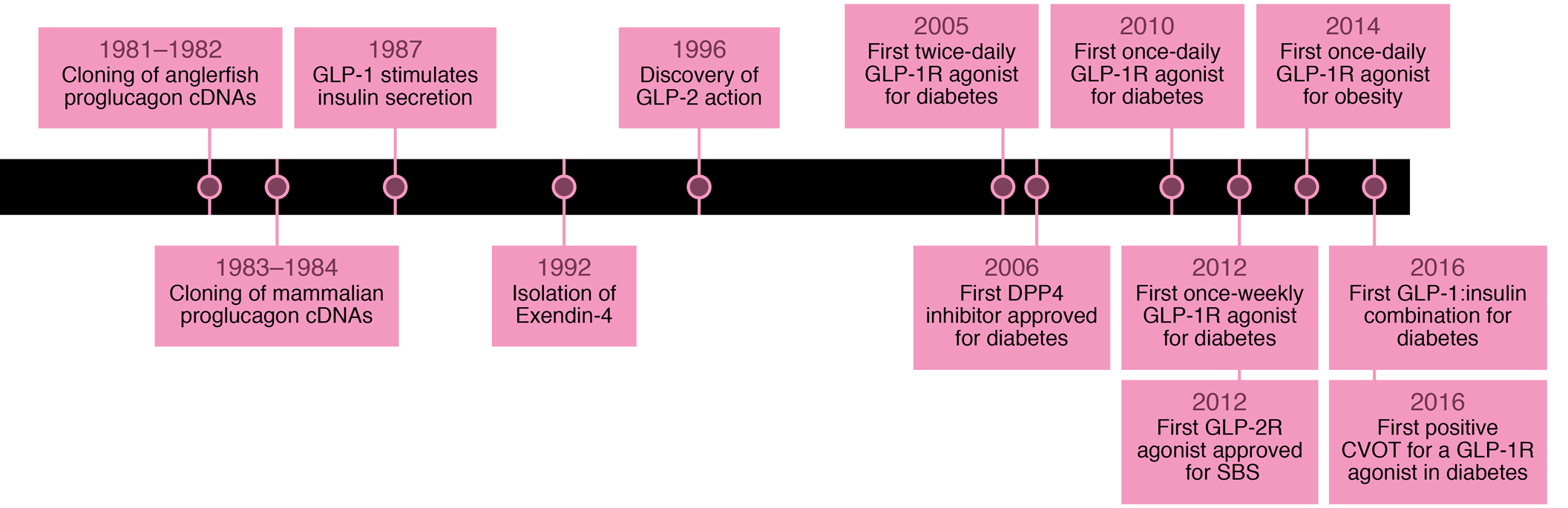
Studi successivi dimostrarono presto (1998) che gli analoghi del GLP-1 resistenti alla DPP-4 avevano un’azione più lunga rispetto al GLP-1 nativo49. Utilizzando inibitori specifici, come il val-pirrolidide, fu possibile inibire completamente la degradazione del terminale NH2 del GLP-1, con immediato incremento della risposta insulinica alla somministrazione orale di glucosio50.
farmaci in commercio
Questo sviluppo fu presto seguito dallo sviluppo di inibitori clinicamente utili del DPP-4, prima vildagliptin (nome commerciale Galvus51) e successivamente sitagliptin52. Sitagliptin fosfato è stato approvato dalla Food and Drug Administration (FDA) statunitense il 17 ottobre 2006 con il nome commerciale Januvia53.
Uno studio clinico randomizzato controllato sul diabete tipo due in 415 soggetti arruolati, sponsorizzato dalla compagnia farmaceutica Novo Nordisk A/S, venne intrapreso nel 200854. Esso confrontava la semaglutide55 con placebo e liraglutide. Semaglutide è un agonista del recettore del GLP-1. Semaglutide è commercializzato in due formulazioni indicate come antidiabetici: Ozempic (siringhe pronte all’uso per iniezione sottocutanea) e Rybelsus (compresse). Una terza formulazione iniettabile per uso sottocutaneo, denominata Wegovy, indicata per il trattamento cronico dell’obesità, è stata successivamente autorizzata nel 2023 per l’immissione in commercio dall’EMA (European Medicines Agency) e dall’Agenzia Italiana del Farmaco (AIFA)56.
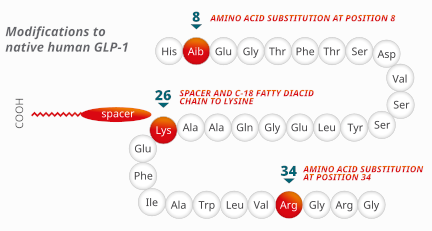
Semaglutide presenta un’omologia di sequenza del 94% rispetto al GLP-1 umano e agisce da agonista per il suo recettore, attivandolo. A differenza del GLP-1, tuttavia, è resistente alla degradazione da parte delle DPP-457.
BIBLIOGRAFIA E RIFERIMENTI:
2 Società Italiana di Diabetologia: “Analoghi di GLP-1”; Divulgazione
3 Bayliss, W. M.; Starling, E. H. (1902). “The mechanism of pancreatic secretion“. J. Physiol. 28 (5): 325–353.
6 “Discovery, characterization, and clinical development of the glucagon-like peptides”, di Daniel J. Drucker, Joel F. Habener and Jens Juul Holst [J Clin Invest. 2017 Dec 1;127(12):4217-4227]
8 Banting FG, Best CH. “Internal secretion of pancreas”. J Lab Clin Med. (1922) 7:251–66
9 “The Origin and Understanding of the Incretin Concept”, by Jens F. Rehfeld [Front Endocrinol. 2018; 9: 387.]
10 La Barre J, Still EU.: “Studies on the physiology of secretin. III. Further studies on the effects of secretin on the blood sugar”. Am J Physiol. (1930) 91:649–53.
12 “The Origin and Understanding of the Incretin Concept”, by Jens F. Rehfeld [Front Endocrinol. 2018; 9: 387.]
15 Yalow RS, Berson SA. “Immunoassay of endogenous plasma insulin in man”. J Clin Invest. (1960) 39:1157–75. 10.1172/JCI104130
16 “Professor Emeritus Neil McIntyre, BSc, M.D., F.R.C.P. (1934–2020)”, di Octavio Campollo
17 “NEW INTERPRETATION OF ORAL GLUCOSE TOLERANCE”, by Neil Mcintyre, M.B. Lond., B.SC., M.R.C.P., C.D Holdsworth, M.B. Leeds, M.R.C.P., D.S Turner, M.SC. Lond., [The Lancet, VOLUME 284, ISSUE 7349, P20-21, JULY 04, 1964]
18 Elrick H, Stimmler L. Hlad CJ, Arai Y. “Plasma insulin responses to oral and intravenous glucose administration”. J Clin Endocrinol Metab. (1964) 24:1076–82.
20 “In Memoriam—John C. Brown, PhD, DSc, FRSC, 1938–2016: Discoverer of GIP and Motilin”, by Timothy J. Kieffer [Gastroenterology, VOLUME 153, ISSUE 5, P1169-1171, NOVEMBER 2017]
21 Brown JC, Mutt V, Pederson RA. “Further purification of a polypeptide demonstrating enterogastrone activity.” J Physiol. (1970) 209:57–64. 10.1113/jphysiol.1970.sp009155
22 Brown JC, Dryburgh JR. “A gastric inhibitory polypeptide. II. The complete amino acid sequence.” Can J Biochem. (1971) 49:867–72.
24 Unger RH, Eisentraut AM, Sims K, McCall S, Madison LL. “Sites of origin of glucagon in dogs and humans.” Clin Res. (1961) 9:53.
25 Unger RH, Ketterer H, Eisentraut AM. “Distribution of immunoassayable glucagon in gastrointestinal tissues.” Metabolism (1966) 15:865–7. 10.1016/0026-0495(66)90156-9
26 Bell GI, Santerre RF, Mullenbach GT. “Hamster preproglucagon contains the sequence of glucagon and two related peptides”. Nature (1983) 302:716–8.
27 Mojsov S, Weir GC, Habener JF. “Insulinotropin: glucagon-like peptide I (7-37) co-encoded in the glucagon gene is a potent stimulator of insulin release in the perfused rat pancreas”. J Clin Invest. (1987)
31 “Her work paved the way for blockbuster obesity drugs. Now, she’s fighting for recognition Svetlana Mojsov helped discover the hormone GLP-1. Why has she been excluded from its history?”. 8 SEP 20235:40 PM ETBYJENNIFER COUZIN-FRANKEL
35 Holst JJ, Orskov C, Nielsen OV, Schwartz TW. “Truncated glucagon-like peptide I, an insulin-releasing hormone from the distal gut.” FEBS Lett. 1987;211(2):169–174.
36 Drucker DJ, Philippe J, Mojsov S, Chick WL, Habener JF. “Glucagon-like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line.” Proc Natl Acad Sci U S A. 1987;84(10):3434–3438
37 Wettergren A, Schjoldager B, Mortensen PE, Myhre J, Christiansen J, Holst JJ. “Truncated GLP-1 (proglucagon 78-107-amide) inhibits gastric and pancreatic functions in man”. Dig Dis Sci. 1993;38(4):665–673.
38 Eng J, Kleinman WA, Singh L, Singh G, Raufman JP. “Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas.” J Biol Chem. 1992;267(11):7402–7405.
40 “The Discovery of GLP-2 and Development of Teduglutide for Short Bowel Syndrome”; by Daniel J. Drucker [ACS Pharmacol. Transl. Sci. 2019, 2, 2, 134–142]
42 “Exenatide once weekly versus twice daily for the treatment of type 2 diabetes: a randomised, open-label, non-inferiority study”, by Daniel J Drucker, John B Buse, Kristin Taylor, David M Kendall, Michael Trautmann, Dongliang Zhuang, Lisa Porter; [Lancet. 2008 Oct 4;372(9645):1240-50.]
44 Deacon CF, Johnsen AH, Holst JJ. “Degradation of glucagon-like peptide-1 by human plasma in vitro yields an N-terminally truncated peptide that is a major endogenous metabolite in vivo.” J Clin Endocrinol Metab. 1995;80(3):952–957.
46 Kameoka J, Tanaka T, Nojima Y, Schlossman SF, Morimoto C (July 1993). “Direct association of adenosine deaminase with a T cell activation antigen, CD26″. Science. 261 (5120): 466–9
47 Hopsu-Havu VK, Glenner GG (1966). “A new dipeptide naphthylamidase hydrolyzing glycyl-prolyl-beta-naphthylamide“. Histochemie. Histochemistry. Histochimie. 7 (3): 197–201.
49 Deacon CF, Knudsen LB, Madsen K, Wiberg FC, Jacobsen O, Holst JJ. “Dipeptidyl peptidase IV resistant analogues of glucagon-like peptide-1 which have extended metabolic stability and improved biological activity.” Diabetologia. 1998;41(3):271–278.
50 “Inhibition of the activity of dipeptidyl-peptidase IV as a treatment for type 2 diabetes.”, by J J Holst; C F Deacon [Diabetes 1998;47(11):1663–1670]
53 “FDA Approves New Treatment for Diabetes”; First in a New Class of Diabetes Drugs