Il tumore del 10%: breve storia
“un rossore frenetico marcato distintamente su ciascuna guancia con un sudore costante, abbondante e generale”
Charles Sugrue
IL FEOCROMOCITOMA
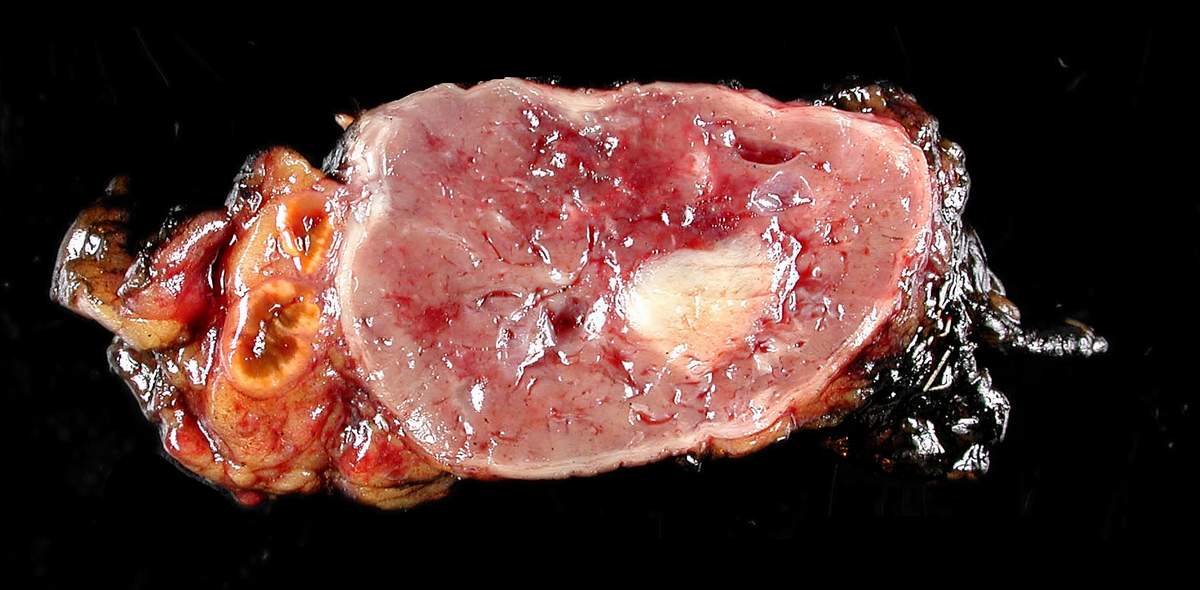
Il feocromocitoma è un tumore raro delle ghiandole surrenali, e origina dalla zona midollare. Fa parte della famiglia dei tumori neuroendocrini. Le cellule neuroendocrine da cui deriva il feocromocitoma sono dette cellule cromaffini e normalmente hanno la funzione di produrre e secernere le catecolamine, in particolare gli ormoni adrenalina e noradrenalina.
{kind=link}
Il feocromocitoma è stato definito il “ten per cent tumor“: nel 10% dei casi insorge nei bambini; nel 10% dei casi è extrasurrenalico; nel 10% dei casi è bilaterale (negli adulti); nel 10% dei casi é multicentrico; nel 10% dei casi insorge come sindrome familiare; nel 10% dei casi è maligno.
ghiandole surrenali

La prima descrizione della ghiandola surrenale risale al 1563 da parte di Eustachio nel libro “Opuscola anatomica“. Nonostante il precedente riconoscimento della presenza delle ghiandole surrenali e della divisione in corteccia e midollo, le osservazioni precise di Addison apparvero nel 1855 secondo cui il ruolo essenziale di queste ghiandole era riconosciuto in pazienti che morivano con distruzione surrenale secondaria alla tubercolosi.
Nel 1800, un medico irlandese, Charles Sugrue (1775-1816) scrisse un caso clinico per il “London Medical and Physical Journal“, descrivendo il caso peculiare di un paziente maschio di otto anni che soffriva di attacchi di dolore apparentemente casuali concentrati nell’addome accompagnati da “un rossore frenetico marcato distintamente su ciascuna guancia” con “un sudore costante, abbondante e generale“. Dopo la morte del piccolo paziente, un gruppo di medici eseguì un’autopsia per determinare la causa della morte e scoprì un tumore oblungo di sei pollici composto da una sconosciuta “sostanza di colore giallastro” proveniente dalla capsula renale (quella che ora è conosciuta come la ghiandola surrenale).
la prima descrizione di un feocromocitoma
 Questa sarebbe diventata la prima descrizione clinica nota di un feocromocitoma, ma poiché non furono descritte le caratteristiche del tumore, il merito completo viene dato al tedesco Felix Fraenkel, che , a Friburgo, fornì un quadro clinico e morfologico di questo tumore in una diciottenne su un articolo del 1886 scritto in tedesco ed intitolato “Un caso di tumore surrenale bilaterale completamente latente e simultanea nefrite con alterazioni del sistema circolatorio e retinite“.
Questa sarebbe diventata la prima descrizione clinica nota di un feocromocitoma, ma poiché non furono descritte le caratteristiche del tumore, il merito completo viene dato al tedesco Felix Fraenkel, che , a Friburgo, fornì un quadro clinico e morfologico di questo tumore in una diciottenne su un articolo del 1886 scritto in tedesco ed intitolato “Un caso di tumore surrenale bilaterale completamente latente e simultanea nefrite con alterazioni del sistema circolatorio e retinite“.
Il resoconto clinico in questione culminava nella morte improvvisa della paziente, coerente con una crisi ipertensiva e un infarto del miocardio. Fraenkel concludeva correttamente che i segni ed i sintomi erano stati causati da un processo in cui
“o una sostanza chimica viene secreta nelle cellule (tumore) e passa nel sangue venoso o in cui le cellule stesse vengono distrutte …”
LUDWIG PICK ED IL FEOCROMOCITOMA
Nel 1912, Ludwig Pick (31agosto 1868 – 3febbraio 1944) coniò il termine “feocromocitoma” dopo aver osservato il consistente cambiamento di colore nei tumori associati al midollo surrenale. La parola feocromocitoma deriva dal greco “phaios” (grigio scuro), “kromos” (colore), “cytos” (cellula) ed il suffisso “oma” (accumulo cellulare).
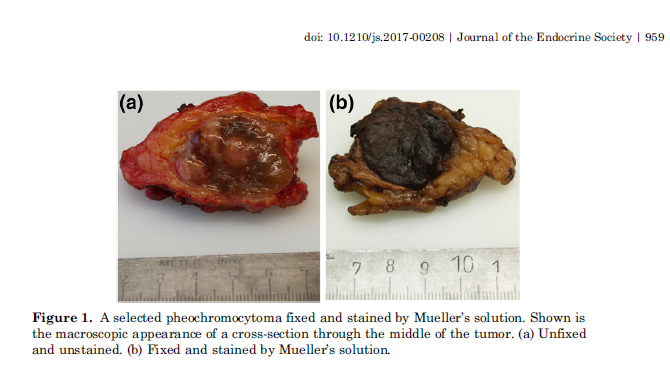
A riguardo, nel 1866, il patologo tedesco Max Schottelius aveva descritto l’aspetto brunastro del tumore dopo esposizione al “fissativo contenente cromato” di Mueller. Questo cambiamento di colore, noto come
“reazione cromaffine“, è determinato dall’ossidazione delle catecolamine e si riflette nel nome feocromocitoma. Così Schottelius eseguiva il primo contributo istochimico noto alla diagnosi, che è diventato lo standard dell’immunoistochimica per la cromogranina (La cromogranina è una glicoproteina, particolarmente conosciuta come marcatore bioumorale specifico per i tumori neuroendocrini).
chirurgia
La prima rimozione chirurgica del feocromocitoma in Europa fu eseguita da Rouks nel 1926 e da Charles Horace Mayo (19luglio 1865 – 26maggio 1939, uno dei fratelli fondatori della “Mayo Clinic”) negli Stati Uniti nel 1927. Il lavoro di Mayo ebbe un impatto sull’evoluzione della conoscenza e trattamento della malattia, anche se Mayo prima dell’intervento non era a conoscenza della diagnosi.
RIFERIMENTI:
- ISSALUTE: Feocromocitoma
- https://it.wikipedia.org/wiki/Feocromocitoma
- https://en.wikipedia.org/wiki/Pheochromocytoma#History
- “Charles Sugrue, M.D., of Cork (1775-1816) and the first description of a classical medical condition: phaeochromocytoma“, di C Cronin (Irish Journal of Medical Science volume 177, pages171–175 – 2008)
- “Trattamento chirurgico del feocromocitoma: esperienza personale“; [Articolo in serbo] di Dejan Elakovic, Dusan Manojlović , Novak Milović (Srp Arh Celok Lek. 2002 lug; 130 Suppl 2:31-7.)
- Fränkel F. “Ein Fall von doppelseitigem, völlig latent verlaufenen Nebennierentumor und gleichzeitiger Nephritis mit Veränderungen am Circulationsapparat und Retinitis“. Arch Pathol Anat Physiol Klin Med. 1886;103:244–263.
- “Max Schottelius: Pioneer in Pheochromocytoma” di Birke Bausch, Arthur S. Tischler, Kurt W. Schmid, Helena Leijon, Charis Eng, Hartmut P. H., Neumann
- https://en.wikipedia.org/wiki/Ludwig_Pick
- http://etimologias.dechile.net/?feocromocitoma
- “Early surgical history of phaeochromocytoma“, di R B Welbourn – British Journal of Surgery, Volume 74, Issue 7, July 1987, Pages 594–596,
- https://en.wikipedia.org/wiki/Charles_Horace_Mayo
- “An Overview of Pheochromocytoma: History, Current Concepts, Vagaries, and Diagnostic Challenges“, di WILLIAM M. MANGER (First published: 07 September 2006)
- http://www.pheochromocytoma.org/earlyhistory/